Курсовая работа: Производство этанола методом гидратации этиленаКурсовая работа: Производство этанола методом гидратации этиленаФЕДЕРАЛЬНОЕ АГЕНТСТВО ПО ОБРАЗОВАНИЮ УФИМСКАЯ ГОСУДАРСТВЕННАЯ АКАДЕМИЯ ЭКОНОМИКИ И СЕРВИСА
Уфимский государственный институт сервиса
Кафедра охраны окружающей среды и рационального использования природных ресурсов
КУРСОВАЯ РАБОТА По дисциплине «Промышленная экология» На тему: Производство этанола методом гидратации этилена
Выполнил студент группы ОД-41 Давыдов Д.В. Проверил профессор Исмагилов Ф.Р. Уфа 2006СОДЕРЖАНИЕ ВВЕДЕНИЕ...................................................................................................3 1. ЭКОЛОГИЗАЦИЯ ХИМИЧЕСКОЙ И НЕФТЕПЕРЕРАБАТЫВАЮЩЕЙ ПРОМЫШЛЕННОСТИ............4 1.1. Теоретические аспекты экологизации и экологизация производства..................................................................................................4 1.2 Очистка отходящих газов.......................................................................5 1.3. Очистка стоков нефтеперерабатывающей промышленности............6 1.4. Выбросы углеводородов........................................................................7 2. ПОДГОТОВКА СЫРЬЯ ДЛЯ ПРОЦЕССА ГИДРАТАЦИИ........8 2.1. Компримирование и осушка газа пиролиза........................................8 2.2. Фракционирование газа пиролиза......................................................10 2.3. Разделение пиролиза при высоком давлении....................................11 2.4. Очистка этилена...................................................................................15 2.5. Получение этилена диспропорционированием пропилена..............18 3. ОСНОВНЫЕ МЕТОДЫ ПОЛУЧЕНИЯ СПИРТОВ......................20 4. ПРОИЗВОДСТВО СПИРТОВ СЕРНОКИСЛОТНОЙ ГИДРАТАЦИЕЙ ОЛЕФИНОВ..............................................................21 4.1. Теоретические сведения......................................................................21 4.2. Технология получения спиртов методом сернокислотной гидратации...................................................................................................28 5. ПРОИЗВОДСТВО СПИРТОВ ПРЯМОЙ ГИДРАТАЦИЕЙ ОЛЕФИНОВ...............................................................................................30 5.1. Теоретические сведения......................................................................30 5.2. Технологические особенности процесса...........................................35 5.3. Технологическая схема процесса.......................................................36 5.4. Характеристика основной аппаратуры..............................................40 5.5. Расчёт материального баланса гидратора..........................................41 6. ПРЯМАЯ ГИДРАТАЦИЯ ЭТИЛЕНА НА НЕЙТРАЛЬНЫХ КАТАЛИЗАТОРАХ..................................................................................46 ЗАКЛЮЧЕНИЕ.........................................................................................47 СПИСОК ИСПОЛЬЗОВАННОЙ ЛИТЕРАТУРЫ.............................48 ВВЕДЕНИЕ В данной курсовой работе исследован процесс получения этанола из этилена путём гидратации последнего. А также рассмотрено получение сырья (этилен) путём фракционирования из газа пиролиза. И последующая его очистка от сероводорода, двуокиси и окиси углерода, ацетилена и кислорода. Этанол принадлежит к числу многотоннажных и широко применяемых продуктов органического синтеза. Он является хорошим, хотя и огнеопасным растворителем; в больших количествах используется в пищевой и медицинской промышленности. Служит горючим в жидкостных ракетных двигателях, антифризом и т. д. Как промежуточный продукт органического синтеза этанол имеет важное значение для получения сложных эфиров: хлороформа, хлораля, диэтилового эфира, ацетальдегида и уксусной кислоты. В России процесс производства синтетического этанола посредством гидратации этилена осуществляется двумя способами: сернокислотной и прямой гидратацией. Первый способ внедрен в промышленном масштабе с 1952г, второй получил широкое распространение в последние десятилетия. На сегодняшний день любой технологический процесс должен рассматриваться не только с точки зрения экономических показателей, но и должно обязательно учитываться влияние этого процесса на окружающую среду. Процесс получения этанола на всех этапах – от подготовки сырья до получения уже готового продукта – сопровождается использованием или образованием опасных для окружающей среды веществ. Таких как серная кислота, фосфорная кислота и др. Совершенно естественно, что выбор метода производства этанола должен учитывать вредное влияние продуктов производства на окружающую среду. Но в то же самое время любой технологический процесс должен быть и экономически целесообразным. Исследование вредного воздействия на окружающую среду с учётом экономической эффективности я и считаю целью данной курсовой работы. 1. ЭКОЛОГИЗАЦИЯ ХИМИЧЕСКОЙ И НЕФТЕПЕРЕРАБАТЫВАЮЩЕЙ ПРОМЫШЛЕННОСТИ 1.1 Теоретические аспекты экологизации и экологизация производства Основные направления инженерной защиты окружающей природной среды от загрязнения и других видов антропогенных воздействий – внедрение ресурсосберегающей, безотходной и малоотходной технологии, биотехнология, утилизация и детоксикация отходов и главное – зкологизация всего производства, при котором обеспечивалось бы включение всех видов взаимодействия с окружающей средой в естественные циклы круговорота веществ. Эти принципиальные направления основаны на цикличности материальных ресурсов и заимствованы у Природы, где, как известно, действуют замкнутые циклические процессы. Технологические процессы, в которых в полной мере учитываются взаимодействия с окружающей средой и приняты меры к предотвращению отрицательных последствий, называют экологизированнными. Подобно любой экологической системе, где вещество и энергия расходуются экономно и отходы одних организмов служат важным условием существования других, производственный экологизированный процесс, управляемый человеком, должен следовать биосферным законам, и в первую очередь закону круговорота веществ. Можно выделить следующие основные направления в осуществлении экологически чистых технологических процессов: 1) комплексное использование и глубокая переработка сырья. Производство должно быть как можно менее ресурсоемким, осуществляться с минимумом затрат сырья и реагентов на единицу продукции. Образующиеся полуфабрикаты должны передаваться в качестве сырья другим производствам и полностью перерабатываться; 2) оптимальное использование энергии и топлива. Производство должно осуществляться при минимальных затратах энергии и топлива на единицу продукции и, следовательно, тепловые загрязнения окружающей среды также минимальны; 3) создание принципиально новых малоотходных технологических процессов. Этого можно добиться совершенствованием катализаторов, техники и технологии производств; 4) создание и внедрение замкнутых систем водоиспользования, включающих (или сводящих к минимум) потребление свежей воды и сброс сточных вод в водоемы; 5) обеспечение высокой эксплуатационной надежности, герметичности и долговечности функционирования оборудования и всех производств. Разработка автоматизированных систем обеспечения экологической безопасности производств и комплексов; 6) обеспечение высокого качества целевых продуктов, используемых в народном хозяйстве. Экологически чистыми должны быть не только сами технологические процессы, но и выпускаемые в них товарные продукты. 1.2. Очистка отходящих газов При производстве этанола выделяются вредные вещества, которые могут загрязнять атмосферу. Такие как диэтилсульфат, этилсульфат, диэтиловый эфир, фосфорные эфиры и другие летучие вещества. Поэтому требуется очистка отходящих газов. Для защиты воздушного бассейна от негативного антропогенного воздействия в виде загрязнения его вредными веществами используют следующие меры: - экологизацию технологических процессов; - очистку газовых выбросов от вредных примесей; - рассеивание газовых выбросов в атмосфере; - устройство санитарно-защитных зон, архитектурно-планировочные решения и др. Наиболее радикальная мера охраны воздушного бассейна от загрязнения - экологизация технологических процессов и прежде всего создание замкнутых технологических циклов, безотходных и малоотходных технологий, исключающих попадание в атмосферу вредных загрязняющих веществ. Экологизация технологических процессов предусматривает создание непрерывных технологических процессов производства, замену местных котельных установок на централизованное тепло, предварительное очищение топлива и сырья от вредных примесей, замену угля и мазута на природный газ, применение гидрообеспылевания, перевод на электропривод компрессоров и др. всё шире применяют частичную рециркуляцию , то есть повторное использование отходящих газов. К сожалению, нынешний уровень развития экологизации технологических процессов, внедрение замкнутых технологий и т. д. недостаточен для полного предотвращения выбросов токсичных веществ в атмосферу. Поэтому на предприятиях повсеместно используются различные методы очистки отходящих газов. Способы очистки выбросов от токсичных газо- и парообразных примесей подразделяют на три группы: 1) поглощение примесей путём применения каталитическ4ого превращения; 2)промывка выбросов растворителями примеси (абсорбционный метод) и 3)поглащение газообразных примесей твёрдыми телами (адсорбционный метод). С помощью каталитического метода токсичные компоненты промышленных выбросов превращают в вещества безвредные или менее вредные путём введения катализаторов. Широко применют палладийсодержащие и ванадийсодержащие катализаторы. Одна из разновидностей этого метода это дожигание вредных примесей с помощью газовых горелок, широко используется на нефтеперерабатывающих заводах. Абсорбционный метод основан на поглощении вредных веществ абсорбентом (вода, растворы щелочей или соды, аммиака и др.) устройства – абсорберы. Адсорбционный метод позволяет извлекать вредные компоненты из промышленных выбросов с помощью адсорбентов – твёрдые тела с ультрамикропористой структурой (активированный уголь и глинозём, силикагель, цеолиты, сланцевая зола и др. ). 1.3. Очистка стоков нефтеперерабатывающей промышленности При производстве этанола образуются стоки фосфорной и серной кислоты. Для очистки используют нейтрализацию.для нейтрализации кислот в сточные воды вводят специальные реагенты (известь, кальцинированную соду, аммиак, раствор едкого натрия). При этом происходит реакция: Н2SO4 + 2NaOH = Na2SO4 + 2H2O H3PO4 + 3NaOH = Na3PO4 + 3H2O 1.4. Выбросы углеводородов Источником загрязнений атмосферы углеводородами является реактор установки каталитического крекинга. Так как смесь газообразных углеводородов является продуктом процесса, то потеря его в атмосферу является ни сколько экологической проблемой для завода НПЗ, сколько экономической. Свойственный деструктивным процессам режим высоких температур и в ряде случаев высокого давления способствует потерям углеводородов и сопутствующего им сероводорода в атмосферу. При этом потери будут в несколько раз больше, чем при низкотемпературных процессах. Степень загрязнения атмосферы углеводородами зависит также от системы охлаждения нефтепродуктов, получаемых на установках каталитического крекинга, и от стабилизации бензиновых фракций. Естественно, что потери от испарения будут тем меньше, чем ниже температура охлаждения продукта, особенно лёгкого бензина. Аналогично будет влиять полнота стабилизации бензина, поскольку газ, растворённый в бензине, повышает парциальное давление углеводородных паров. Поэтому для предотвращения потери продукта, газы идущие из реактора охлаждают и сконденсировавшиеся жидкие продукты направляют на колонну стабилизации. Углеводороды, которые идут из регенератора установки не требуют дополнительной отчистки, так как их имеется незначительное количество, и они окисляются до CO2 и H2O в дожигателе CO. [1] 2. ПОДГОТОВКА СЫРЬЯ ДЛЯ ПРОЦЕССА ГИДРАТАЦИИ 2.1.Компримирование и осушка газа пиролиза Этилен выделяют из газа пиролиза при низких температурах и высоких давлениях. Перед фракционированием газ компримируют до давления 34 – 45 кгс/см2. Компримирование производится во избежание перегрева газа при фракционировании, что привело бы к полимеризации диенов и высших олефинов. Осушка необходима потому, что газообразные углеводороды при низких температурах и высоком давлении образуют с водой гидраты – кристаллические комплексы типа СН4. 6Н2О, С2Н6. 7Н2О и так далее. Кристаллогидраты затрудняют транспортирование газа, а при фракционировании, выделении гидратов и льда может вызывать забивание аппаратуры и нарушение нормальной работы газофракционирующей установки. Компримирование газа пиролиза производится в трёх -, четырёх- или пятиступенчатых компрессорах с промежуточным охлаждением и сепарацией газа между ступенями. Для этой цели могут применяться поршневые компрессоры или турбокомпрессоры (последние более экономичные и надёжные). Очень важно обеспечить эффективное межступенчатое охлаждение газа до низких температур. Для этого применяют поверхностное охлаждение. Показана также возможность проводить охлаждение газа прямым контактом его хладоагентом. Как известно, температура газа после сжатия зависит от степени сжатия и, следовательно, она тем ниже, чем больше число ступеней сжатия. При четырёхступенчатом сжатии до 45 кгс/см2 температуры газа на выходе из компрессора не выше 100°C. В межступенчатых холодильниках конденсируется вода и тяжёлые углеводороды, которые должны быть тщательно отделены от газа. Содержание водяных паров в газе (кг/кг) зависит от температуры и давления:
где Z – количество влаги, кг/час; G – количество газа, кг/час; Р – давление паров воды при температуре газа, кгс/см2 ; π – давление в системе, кгс/см2; М – средний молекулярный вес газа. При повышении давления и понижении температуры часть водяных паров конденсируется, что и происходит при компрессии с последующим охлаждением. Поэтому осушку газа проводят после компрессии, чтобы на осушку поступал газ с наименьшим содержанием влаги. Для надёжной работы газофракционирующих установок точка росы газа пиролиза не должна превышать -65°C, что отвечает содержанию в нём воды при 43 кгс/см2 менее 30 мг/кг. Для осушки газа могут применяться жидкие реагенты (ди- или триэтиленгликоли) и твёрдые адсорбенты, однако для осушки жидкими реагентами требуется более сложная аппаратура и она менее эффективна, чем адсорбционная. В качестве адсорбентов применяют твёрдые материалы с развитой поверхностью – силикагель, алюмогель, природные алюмосиликаты и синтетические цеолиты (алюмосиликаты натрия и кальция с регулируемым размером пор, так называемы молекулярные сита). Молекулярные сита селективно адсорбируют молекулы определённого размера; их поглотительная способность в 3 – 4 раза выше, чем у алюмогелей и силикагелей, благодаря чему значительно уменьшаются размеры осушителя. Таким образом применение молекулярных сит позволяет снизить влажность газа на целый порядок по сравнению с алюмогелем и силикагелем; в результате точка росы газа может быть снижена до -73°C. Перед осушкой газ освобождают от тяжёлых углеводородов для чего его после компримирования, охлаждения и сепарации подвергают ректификации. Отделение тяжёлых углеводородов необходимо во избежание забивания пор адсорбента и его дезактивации, а также потому, что на разделение очень важно подавать газ, свободный от тяжёлых фракций. Перед осушкой газ должен быть охлаждён, так как с понижением температуры увеличивает влагоёмкость адсорбента. Осушка газа проводится в периодически работающих колоннах; циклы работы колонн 24 – 48 ч.
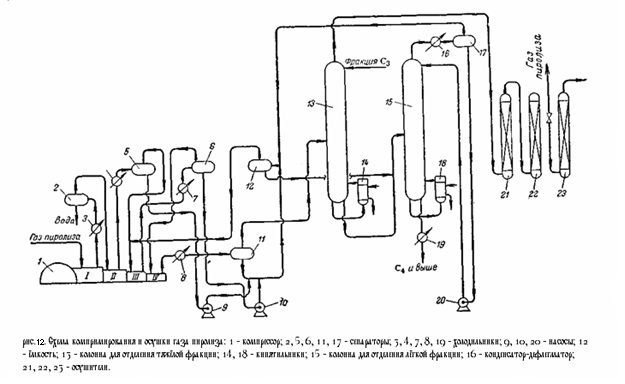
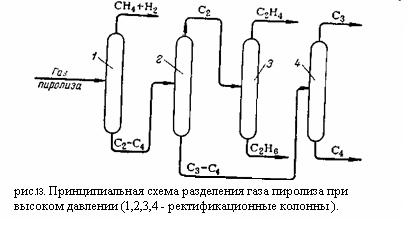
На (рис.12) приведена технологическая схема компримирования и осушки газа пиролиза. Газ из цеха пиролиза забирается компрессором 1 и проходит последовательно все ступени компрессии. После каждой ступени газ охлаждается в межступенчатых холодильниках 3, 4 и 7 и отделяется от конденсата в межступенчатых сепараторах 2, 5, 6. После четвёртой ступени компремированый газ охлаждается в холодильнике 8 до 15 °C, отделяется от конденсата в сепараторе 11 и направляется в колонну 13 для выделения тяжёлых углеводородов (С4 и выше). Конденсат из сепаратора 1 направляется в ёмкость 12, куда поступают также конденсаты из многоступенчатых сепараторов. Колонна 13 снабжена кипятильником 14, обогреваемым водяным паром, и орошается жидкой пропан-пропиленовой фракцией. Температура вверху колонны 0 °C, внизу 85 – 90 °C, давление 45 кгс/см2 . Для предотвращения полимеризации в колонну вводится ингибитор. Газ, отбираемый с верха колонны, направляется на осушку. В кубовой жидкости колонны 13 остаётся значительное количество лёгких углеводородов, в том числе этилен и пропилен. Для их выделения кубовая жидкость направляется в колонну 15 орошаемую конденсатом из ёмкости 12. Колонна снабжена кипятильником. Температура вверху колонны 32 °C, внизу 125 °C. Верхний продукт конденсируется в охлаждаемом водой конденсаторе 16, отделяется от газа в сепараторе 17, и конденсат в виде флегмы возвращается на орошение. Газ из ёмкости 12 присоединяется к сырью на третьей ступени компрессии. Кубовая жидкость (С4 и выше) охлаждается в холодильнике 19 и выводится из системы. Осушка газа из колонны 13 осуществляется последовательно в трёх осушителях 21, 22, 23, заполненных адсорбентом. Поочерёдно в двух из них происходит осушка газа, а в третьем – регенерация адсорбента. Регенерация заключается в продувке адсорбента инертным газом – в данном случае метано-водородной фракцией, нагретой до 240 °C. Через каждые 24 часа работы первый по ходу газа осушитель отключается для регенерации, второй становится первым, а осушитель со свежерегенерированным адсорбентом становится вторым по ходу газа. Приведённая схема является одним из возможных вариантов компримирования газа пиролиза и отделения тяжёлых фракций. Иногда тяжёлые фракции отделяют перед последней ступенью компрессии. Существует также схемы, предусматривающие выделение тяжёлых фракций, как перед осушкой газа, так и перед газоразделением, с отделением части пропан-пропиленовой фракции на последней ступени компримирования. 2.2.Фракционирование газа пиролиза Для разделения газа пиролиза применяют следующие методы. 1. конденсационно-ректификационный метод (низкотемпературная ректификация), когда разделение газовой смеси – деметанизация, выделение и разделение этан-этиленовой фракции – достигается конденсацией с последующей ректификацией под давлением с применением аммиачного, метанового, этиленового (или пропанового) холодильных циклов. 2. абсорбционно-ректификационный метод, при котором все компоненты тяжелее метана извлекают из газа абсорбцией при низких температурах и затем выделяют низкотемпературной ректификацией. Оба метода требуют затрат холода и применения специальных хладогентов, поскольку критическая температура этилена равна +9,7 °C и ожижение его водой невозможно. Необходимо отметить, что при конденсационно-ректификационном методе основные затраты приходятся на создание низких температур. В связи с этим большое значение имеет эффективность и экономичность применяемых холодильных циклов. Разделение газа пиролиза может осуществляться при низком или при высоком давлении. При разделении при низком давлении (температура ниже -120 °C, давление 1,3 – 2 кгс/см2 ) расширяется интервал температур кипения разделяемых углеводородов и увеличивается их относительная летучесть. Кривая равновесия фаз становится круче, вследствие чего для разделения требуется меньше тарелок, флегмовое число снижается, а чёткость разделения может быть очень высокой. С повышением давления кривая равновесия фаз становится более пологой – увеличивается число тарелок и флегмовое число. Однако для создания низких температур, требуемых для разделения при низком давлении, приходится применять наряду с аммиачным и пропановым также метановый холодильный цикл. Это требует более сложного оборудования и менее экономично, чем этиленовый холодильный цикл, применяемый при высоких давлениях. Вместе с тем, хотя на установках газоразделения при низком давлении получается очень чистый этилен, они малопроизводительны и очень чувствительны к изменению состава газа. Кроме того, их значительно труднее автоматизировать, чем установки высокого давления. 2.3.Разделение пиролиза при высоком давлении При высоком давлении разделение может производиться абсорбционно-ректификационным или конденсационно-ректификационным методами. При использовании конденсационно-ректификационного метода метано-водородная фракция выделяется при температурах от -90 до -100°C, при абсорбционно-ректификационном методе – от -20 до -30°C с использованием лёгкого абсорбента типа фракции С4. Принципиальная схема разделения газа пиролиза при высоком давлении приведена на рисунке 2:
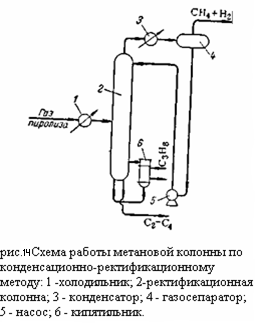
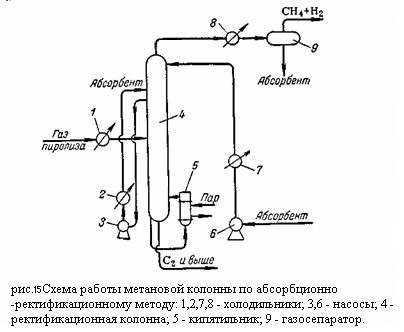
Компримированный, осушенный и охлаждённый газ поступает в метановую колонну 1, где из него выделяются газообразные метан и водород, которые отводятся сверху колонны. Углеводороды С2 – С4 конденсируются в колонне 1 и направляются в этан-этиленовую колонну 2. С верха этой колонны отбирается этан-этиленовая фракция направляемая в этиленовую колонну 3, где этилен и этан разделяется: сверху отбирается концентрированный этилен, а снизу этан. Остаток из колонны 2 представляющий собой смесь из углеводородов С3 – С4, направляется в пропан-пропиленовую колонну 4. сверху из этой колонны отбирается пропан пропиленовая фракция, а снизу бутан-бутеновая. При разделении газа пиролиза при высоком давлении конденсационно-ректификационный и абсорбционно-ректификационный методы различаются в основном лишь схемой и режимами работы метановой колонны. Схема работы метановой колонны при конденсационно-ректификационном методе изображена на рисунке 3:
Газ пиролиза в холодильнике 1 охлаждается до минус 55 – 60 градусов Цельсия с использованием аммиачного или пропанового холодильного цикла и поступает в ректификационную колонну 2. Из верхней части колонны 2 (температура вверху минус 95 – 100 градусов Цельсия) отбираются пары метано-водородной фракции, проходящий через охлаждаемый этиленом конденсатор 3, в котором конденсируется часть метана, требуемая для орошения колонны. Конденсат отделяется от паров в газосепараторе 4 и насосом 5 подаётся на орошение колонны 2. Нижняя часть колонны 2 обогревается пропаном с помощью кипятильника 6 (температура внизу колонны 15 – 18 градусов Цельсия). Снизу из колонны выводится смесь углеводородов (С2 и выше), направляемая на дальнейшую ректификацию. При абсорбционно-ректификационном методе метановая колонна (рис.15):
Представляет собой фракционирующий абсорбер (верх колонны работает как абсорбер, а низ – как отпарная колонна). Газ пиролиза перед поступлением в колонну 4 охлаждается в холодильнике 1 до -25°С. Извлечение компонентов тяжелее метана осуществляется путём орашения колонны 4 лёгким абсорбентом (фракция С4 из пропановой колонны), также охлаждённым до -25°С. Расход абсорбента достигает 1,35 кг на кг газа. Необходимость работать с лёгким абсорбентом обусловлена тем, что степень извлечения (абсорбционный фактор) пропорционален числу молей абсорбента: где A – абсорбционный фактор; K – константа фазового равновесия извлекаемого компонента; L, G – количество абсорбента и газа, кмоль. Следовательно, при той же массе абсорбента степень извлечения будет больше для абсорбента с более низким молекулярным весом. Кроме того, для десорбции более лёгкого абсорбента требуются меньшая затрата тепла вследствие более низкой температуры низа десорбера. Недостатком применения лёгких абсорбентов является частичный их унос с отходящими газами, в данном случае с метано-водородными фракциями. Для отвода тепла, выделяющегося при абсорбции, верхняя часть колонны 4 оборудована промежуточными выносными холодильниками 2. уходящая сверху метано-водородная фракция содержит некоторое количество паров абсорбента, зависящее от температуры и давления на верхней тарелке. Для извлечения унесённого абсорбента метано-водородная фракция охлаждается в холодильнике 8 до -60 °С и поступает в ёмкость 9, где газ отделяется от конденсата. Обогрев низа колонны осуществляется через кипятильник 5. однако, поскольку остаток содержит значительное количество абсорбента (фракция С4), температура низа колонны должна быть выше, чем в отсутствии абсорбента, и составляет около 60°C. Соответственно обогрев кипятильника осуществляется водяным паром. Схемы работы колонн 3 и 4 (рисунок 20) такие же, как при конденсационно-ректификационном методе. В режиме этан-этиленовой колонны 2 имеются различия обусловлены большим содержанием абсорбента фракции С4 в остатке. Температура низа этан-этиленовой колонны при работе по конденсационно-ректификационному методу должна быть около 70 °C, в то время как при абсорбционно-ректификационном методе она повышается до 110°C. Соответственно для обогоева кипятильников требуется в первом случае пар низкого, а во втором – высокого давления, при чём расход водяного пара для абсорбционно-ректификационной схемы значительно больше, так как абсорбент циркулирует через все колонны (за исключением этиленовой). Больше также расход воды на охлаждение пропан-пропиленовой колонны. Чистота этилена также получается различной при работе по разным схемам. При одинаковой чёткости разделения в отгонной части метановой колонны абсолютное содержание метана в остатке при работе по схеме с абсорбции будет больше, вследствие того, что количество остатка (фракция С2 – С4 и абсорбент) примерно в три раза больше. Поэтому содержание метана в этилене, полученном абсорбционно-ректификационным методом больше, чем при работе по схеме с конденсацией. Резюмируя, отметим преимущества и недостатки каждого метода. Преимуществами конденсационно-ректификационного метода является меньший расход водяного пара и воды и большая чистота этилена, недостатками – сложность компрессорного оборудования, более низкие температуры и высокие требования к стабильности состава газа. К достоинствам абсорбционно-ректификационного метода относятся умеренно низкие температуры и сравнительная простота эксплуатации; к недостаткам – повышенные энергетические затраты, унос абсорбента и необходимость его выделение при пониженных температурах. По суммарным технико-экономическим показателям предпочтение следует отдать конденсационно-ректификационному методу. Технико-экономические показатели процесса низкотемпературного разделения газа пиролиза определяются, в первую очередь, энергетическими затратами на создание низких температур, а эти затраты в значительной мере зависят от выбранной схемы охлаждения. Охлаждения до низких температур, необходимое для выделения из газа пиролиза, достигается сочетанием методов дросселирования, расширения газа в детандере и каскадного охлаждения за счёт теплообмена с испаряющимся вышекипящим компонентом (например, охлаждение этилена пропаном, метана – этиленом). 2.4. Очистка этилена Очистка этилена сводится к удалению из него сероводорода, двуокиси и окиси углерода, ацетилена и кислорода. Удаление этих примесей может осуществляться на различных стадиях процесса производства этилена. Сероводород, двуокись углерода и органические сернистые загрязнения удаляют путём промывки газо-водной щёлочью (обычно 10% раствором едкого натра) в специальном скруббере. При этом протекают реакции: H2S + 2NaOH → Na2S + 2H2O CO2 + 2NaOH → Na2CO3 + H2O COS + 4NaOH → Na2CO3 + Na2S+ 2H2O CS2 + 6NaOH → Na2CO3+ 2Na2S+ 3H2O Значительное снижение содержания сернистых соединений (до 0,0001%) и двуокиси углерода (до 0,001%) достигается при двухступенчатой промывке щёлочью. Очистка от ацетилена может осуществляться промывкой ацетоном при низких температурах либо, чаще всего, селективным гидрированием. Содержание ацетилена в газе пиролиза колеблется от 0,1 до 1%. При очистке оно должно быть снижено до 0,001 – 0,002 %. Удаление ацетилена абсорбции ацетоном основано на предпочтительном растворении ацетилена в ацетоне и проводится при низких температурах. Охлажденный этилен промывают ацетоном в абсорбционной колонне 2; насыщенный ацетон регенерируется в колонне 8 путём отгона ацетилена и после охлаждения в системе теплообменников и холодильников вновь поступает в абсорбционную колонну 2. Очистка от ацетилена селективным гидрированием основана на реакции: С2Н2 + Н2 → С2Н4 Условия процесса выбираются таким образом, чтобы практически избежать побочной реакции гидрирования этилена. При гидрировании этилен также освобождается от кислорода и от окиси углерода: О2 + 2Н2 → 2Н2О СО + 3Н2 → СН4 + Н2О В качестве катализаторов гидрирования могут применяться никель на кизельгуре, палладий на активированном угле или платина на окиси алюминия. В качестве водосодержащего газа для гидрирования применяется метано-водородная фракция. Гидрирование может проводиться при давлениях 20 - 30 кгс/см2 или более низких и при температуре газа на входе в реактор от 100 до 180 – 200 °C. Избирательное гидрирование ацетилена может проводиться в реакторах трубчатых или колонного типа. В первом случае тепло реакции отводится циркулирующим в межтрубном пространстве сырьём или водой, во втором поддувом холодного гидрированного продукта:
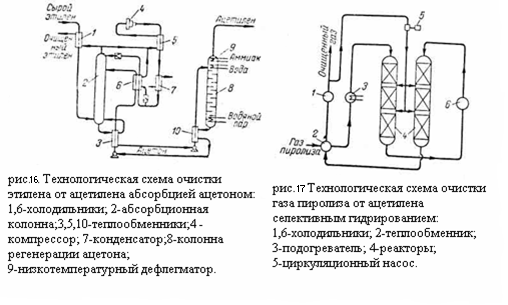
На рисунке приведена технологическая схема очистка газа пиролиза от ацетилена селективным гидрированием. Газ пиролиза после компримирования и осушки проходит теплообменник 2 и паровой подогреватель 3 с температурой 150 - 200°C проходит последовательно реакторы 4 колонного типа. Очищенный газ пиролиза через теплообменник 2 и холодильник 1 направляется на дальнейшее разделение. Часть очищенного газа циркуляционным компрессором 5 подаётся в реакторы 4 для снятия теплоты реакции. Если очистке гидрированием подвергается этиленовая фракция, после гидрирования необходимо концентрирование этилена. Принципиальная технологическая схема доочистки и концентрирования этилена приведена на (рис.18):
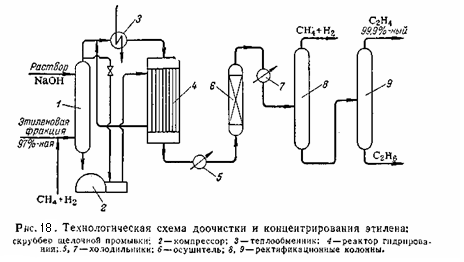
Этиленовая фракция (97 – 98 % этилена) смешивается с метано-водородной фракцией и поступает в скруббер 1 для промывки раствором щёлочи с целью удаления углекислого газа, сероводорода и органических сернистых соединений. Отработанная щёлочь выводится снизу, а газ сверху. Газ после щелочной промывки делится на два потока. Один поток проходит межтрубное пространство реактора гидрирования 4 и смешивается со вторым холодным потоком перед паровым подогревателем 3. в паровом подогревателе 3 газ нагревается до 100 – 190 °C и направляется в реактор 4 на гидрирование. Реактор представляет собой трубчатый аппарат, в трубки которого загружен катализатор, например, палладий на активированном угле. Тепло реакции отводится холодной этиленовой фракцией циркулирующей через межтрубное пространство. Давление в реакторе около 23 кгс/см2. После гидрирования газ охлаждается в холодильнике 5 и направляется в осушитель 6 для удаления влаги. Осушенный газ после дополнительного охлаждения в холодильнике 7 направляется в колонну 8 и 9. Сверху из колонны 8 при температуре -38°C отбирается метано-водородная фракция с содержанием этилена до 70%, который возвращается на компримирование. Остаток колонны 8 освобождается от этана в колонне 9. Сверху из колонны 9 при температуре -28°C отбирается 99,9%-ный этилен. Остаток колонны 9 содержит до 60% этана. Температура низа колонны 9 около -20 °C. 2.5. Получение этилена диспропорционированием пропилена Пропилен является побочным продуктом процесса пиролиза на этилен. В связи с этим разработан так называемы процесс «триолефин»,основанная на реакции диспропорционирования пропилена: 2 С3Н6 ↔ С2Н4 + СН3СН=СНСН3
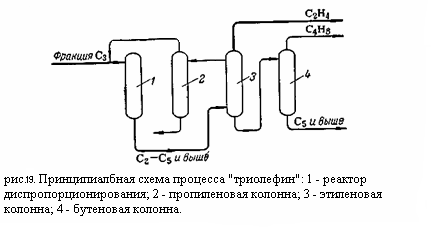
Реакция идёт на окисных катализаторах (Со – Мо или W); в качестве носителя используется окись алюминия. Побочными реакциями являются изомеризация, крекинг и уплотнение: СН3СН=СНСН3 ↔ СН2=СНСН2СН3
СН3СН=СНСН3 + СН2=СНСН2СН3 → Продукты уплотнения При 500 К равновесная глубина превращения пропилена составляет 45,5 мол.%. Основной причиной понижения выхода в реакции диспропорционирования являются нежелательные реакции изомеризации. Реакции крекинга и уплотнения удаётся подавить подбором условий реакции. Исходный пропилен должен быть очищен от воды, сероводорода, кислорода, кислородсодержащих соединений, метилацетилена и пропадиена. Процесс ведут при 66 – 260 °C, давлении 14 – 41 кгс/см2 и высокой скорости подачи сырья. Реактор периодически останавливают для выжигания кокса, отлагающегося на поверхности катализатора. Количество образующегося кокса составляет всего 0,02 масс.% в расчёте на сырьё, но вследствие большой скорости подачи сырья оно достигает 20% от массы катализатора. Цикл работы между регенерациями колеблется от 20 часов до нескольких суток. Диспропорционирование идёт с большой избирательностью: сумма этилена и бутенов достигает 95 – 97 % от превращённого пропилена, а конверсия пропилена 40 – 45 %. Непрореагировавший пропилен возвращается в реактор. Мольное отношение этилена к бутенам близко к стехиометрическому (около 1:1). Бутен используется для дегидрирования с получением бутадиена-1,3. Принципиальная схема процесса «триолефин» изображена на (рис.19): Пропан-пропиленовая фракция С3 в смеси с рециркулирующим пропиленом поступает в реактор 1 на диспропорционирование. Продукты реакции поступают в нижнюю часть этиленовой колонны 3 из которой сверху отбирается этилен и в виде бокового погона – смесь пропана с пропиленом. Эта смесь направляется в пропиленовую колонну 2, где непревращённый пропилен отгоняется от пропана и возвращается в рецикл. Остаток колонны 3 поступает в бутеновую колонну 4 для отделения высококипящих углеводородов. Из верхней части колонны 4 отбираются бутены высокой чистоты. Процесс «триолефин» позволяет увеличить выход этилена при пиролизе бензина с 26 – 30 до 40,9 % и бутадиена – с 4,7 до 5,7 % при одновременном снижении выхода пропилена с 17,5 до 1,5% и бутенов с 3,5 до 0,4%. [3] 3. ОСНОВНЫЕ МЕТОДЫ ПОЛУЧЕНИЯ СПИРТОВ Спирты применяют в производстве синтетических полимеров, каучуков, пластификаторов, моющих средств, в качестве растворителей и экстрагентов и для других целей. Они являются массовой продукцией нефтехимического синтеза, поэтому большое значение для экономики производства спиртов имеют методы их получения и исходное сырьё. Одним из важнейших методов производства спиртов является гидратация олефинов. Этим методом получают этиловый, изопропиловый, втор- и трет-бутиловые спирты. Метиловый спирт получают на основе окиси углерода и водорода. Наиболее крупнотоннажным продуктом является этиловый спирт. На основе этилового спирта в конце тридцатых годов С.В. Лебедевым был разработан метод получения бутадиена-1,3 и синтетического каучука на его основе. Этиловый спирт получают из пищевого сырья (ферментативный метод), из продуктов гидролиза древесины, из сульфитных щелоков и этилена. Трудовые и сырьевые затраты при производстве этилового спирта из пищевых продуктов и древесных опилок очень велики, поэтому значительно выгоднее исходить из дешёвого углеводородного сырья и получать спирт гидратацией этилена. Для производства одной тонны этилового спирта на основе этилена необходимо переработать всего примерно 2,5 тонны газа или нефтяных дистиллятов, а для получения 1 тонны спирта из растительных материалов требуется 4 тонны зерна, 10 – 12 тонн картофеля или 8 тонн древесных опилок. Трудовые затраты в человеко-часах при производстве этанола из разных источников составляют: из картофеля 280, из зерна 160, из этилена 10. Для получения синтетического этанола сырьём служит этилен, который подвергают сернокислотной гидратации или гидратации на твёрдых фосфорно-кислотных катализаторах (прямая гидратация): С2Н4 + Н2SO4 → C2H5OSO3H + H2O → C2H5OH + Н2SO4; С2Н4 + H2O (H3PO4) → C2H5OH. Себестоимость синтетического этилового спирта значительно ниже, чем при других методах его получения. Относительная себестоимость (в %) этанола, получаемого разными способами, составляет: из этилена 100%, гидролизом древесины 174 – 290, из пищевого сырья 300 – 400. [2] 4. ПРОИЗВОДСТВО СПИРТОВ СЕРНОКИСЛОТНОЙ ГИДРАТАЦИЕЙ ОЛЕФИНОВ 4.1. Теоретические сведения Реакция присоединения воды была открыта Фарадеем 1825 – 1828 гг. он нашёл, что при действии серной кислоты на этилен, содержащийся в светильном газе, наряду с диэтиловым эфиром и другими продуктами образуется этиловый спирт. Впоследствии было установлено, что первым продуктом присоединения серной кислоты к этилену является этилсерная кислота, которая при гидролизе превращается в этанол. В 1873 году А.М.Бутлеров и В.Горяинов детально изучили сернокислотную гидратацию этилена и предсказали техническое значение этого процесса. В начале тридцатых годов в Советском Союзе М.А.Далиным с сотр. были проведены исследования сернокислотной гидратации олефинов и в 1936 году в Баку была создана первая промышленная установка по получению этилового спирта из нефтяных газов. Сернокислотная гидратация олефинов является обратимым процессом. Она протекает в две стадии: CH2=CH2 + Н2SO4 ↔ CH2OSO2OHCH3 + H2O ↔ CH2OHCH3+ Н2SO4
Первая стадия – взаимодействие олефинов с серной кислотой – протекает через образование карбоний-иона, то есть как электрофильное замещение по правилу Марковникова. Поэтому сернокислотная гидратация олефинов выше С2 позволяет получать только вторичные и третичные спирты. Серная кислота в этом процессе играет роль и катализатора и реагента. Сначала происходит отщепление протона от молекулы кислоты: Н2SO4 ↔ Н+ + -OSO2OH Под действием его из молекулы олефина образуется карбоний ион CH2=CH2 + Н+ → CH2+CH3
который далее реагирует с серной кислотой с отщеплением от неё протона и образованием алкилсульфатов: CH2+CH3 +Н2SO4 ↔ CH2OSO2OHCH3 + Н+
Если в системе присутствует вода, могут также образовываться ионы алкоксония, которые разлагаются с образов()анием спирта: CH2+CH3 + H2O ↔ CH2(H2O) +CH3 → C2H5OH + Н+
Наряду с этим протекает ряд побочных реакций: а)образование диалкил сульфатов:
CH2OSO2OHCH3 + CH2=CH2 → (CH3CH2)2SO4 + Н2SO4
б) образование простых эфиров из двух молекул спирта с отщеплением воды: 2C2H5OH +Н2SO4
Причём предполагается, что фактически сначала спирт реагирует с карбоний-ионом, а потом от продукта присоединения отщепляется протон: C2H5OH + CH2 +CH3 в) образование карбонильных соединений (альдегидов) при дегидрировании спирта:
C2H5OH г) полимеризация олефинов: nCH2=CH2
Из-за этих побочных реакций при гидратации олефинов наряду со спиртами получаются небольшие количества эфиров, альдегидов и полимеров. Кроме того, образование нерасщепляющихся сульфопроизводных приводит к повышенному расходу серной кислоты. Наиболее низкой реакционной способностью при взаимодействии с серной кислотой обладает этилен. Относительная скорость поглощения разных олефинов 80% серной кислотой меняется следующим образом: этилен (1), пропилен (500), бутилен-1(1 000), изобутилен (16 000). Видно, что с увеличением молекулярного веса олефинов их реакционная способность возрастает. Олефины изостроения также обладают очень высокой реакционной способностью. Поскольку олефины в зависимости от молекулярного веса и строения реагируют с серной кислотой с разной скоростью, для каждого из них подбирают свои условия: концентрация кислоты, температуру, давление.
Абсорбцию олефинов серной кислотой осуществляют в реакторах колонного типа с колпачковыми тарелками, на которых расположены змеевики водяного охлаждения, поскольку реакция идёт с выделением тепла. Тепло выделяется не только за счёт собственной реакции, но также за счёт разбавления кислоты водой. Вторая стадия – гидролиз алкилсульфатов водой, осуществляемый при нагревании острым паром; одновременно происходит отгонка спирта и разбавление серной кислоты до концентрации почти вдвое меньшей, чем исходная. Существенной особенностью процесса является расщепление при гидролизе на спирте кислоту, не только моноалкил-, но и диалкилсульфатов: CH2OSO2OHCH3 + H2O ↔ C2H5OH + Н2SO4
(CH3CH2)2SO4 + 2H2O ↔ 2C2H5OH + Н2SO4 При избытке олефина количество диалкилсульфата возрастает, а расход кислоты снижается, что очень важно для экономики процесса. Обычно один моль серной кислоты поглощает до 1,2 – 1,3 моль олефина. Другой особенностью является возможность поглощения олефинов из соответствующих фракций (этан – этиленовые, пропан – пропиленовые и др.) без их концентрирования. Селективность превращения олефина в спирт при сернокислотной гидратации составляет 85 – 95%, а общая степень конверсии олефина превышает 97%. Олигомеризация этилена При гидратации олефинов наряду с основной реакцией протекают олигомеризация олефина (получение низкомолекулярных полимеров) и образование простого эфира. Все они идут через промежуточную стадию карбокатиона, что можно изобразить схемой: Н2О + R+; -H+(k1) ↔ ROH + R+; -H+(k2) ↔ ROR, R+ + олефин
Для состояния системы, далёкого от равновесия, из этой схемы вытекает следующее уравнение дифференциальной избирательности:
Из него ясно видно что избирательность растёт при наличии избытка воды по отношении к олефину и образующемуся спирту. Спирт более реакционноспособен, чем вода [ (k2/k1)>1], поэтому реакцию надо вести так чтобы сохранялся большой избыток воды по отношению к спирту (~ 15:1). Выход олигомеров зависит от способности олефинов к полимеризации (изобутен > пропилен > этилен). Образование олигомеров можно снизить, не только изменяя соотношение воды и олефина, но и уменьшая температуру, так как олигомеризация имеет более высокую энергию активации по сравнению с гидратацией. Следует отметить, что при приближении к равновесию скорость гидратации и избирательность падают, что делает невыгодным проведение реакции до степеней конверсии, близким к равновесным. При этом для каждого олефина и катализатора имеется некоторый оптимум соотношения реагентов, степени конверсии и температуры, зависящий от производительности и избирательности процесса. Для дегидратации спиртов установлена схема превращения, по которой эфир способен к разложению на олефин и спирт: этанол + H+; – Н2О ↔ R+ + H+ → олефин; этанол + H+; – Н2О ↔ R+ + этанол ↔ ROR + H+
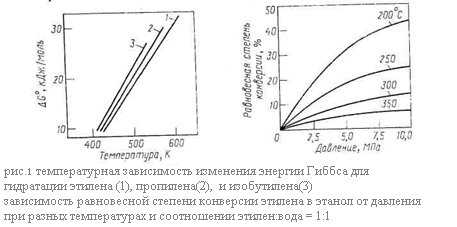
→ ROH + олефин + H+. Внутримолекулярная дегидратация имеет более высокую энергию активации по сравнению с образованием простого эфира. По этой причине, а также из рассмотрения приведённой выше схемы следует, что дегидратацию с образованием ненасыщенной связи надо осуществлять при повышенной температуре и низком парциальном давлении или концентрации спирта. Дегидратацию с образованием простого эфира проводят при более низкой температуре, более высоких концентрации и парциальном давлении спирта (например, под некоторым давлением) и при неполной конверсии спирта в реакторе. В результате рассматриваемых реакций нередко образуется ещё один побочный продукт – альдегид или кетон, получающийся за счёт дегидрирования спирта: С2Н5ОН → СН3СНО. Протонные кислоты не катализируют эту реакцию, но она становится возможной при использовании некоторых носителей или оксидных катализаторов. Из последних наиболее избирательны к дегидратации (по сравнению с дегидрированием) ThO2 и Al2O3, в то время как многие оксиды обладают смешанным, а другие – преимущественно дегидрирующим действием. Термодинамика реакций Рассмотрим равновесие основной реакции: гидратации – внутримолекулярной гидратации. Она протекает с выделением тепла, следовательно её равновесие смещается вправо при понижении температуры. Дегидратации, наоборот, способствует нагревание. Изменение энергии Гиббса при гидратации этилена, пропилена и изобутилена в зависимости от температуры представлено графически на (рис. 1.) Видно, что равновесие невыгодно для гидратации олефинов, так как при 150-300*С, когда катализаторы процесса достаточно активны, энергия Гиббса имеет большую положительную величину и равновесие смещается в сторону дегидратации. При этом для олефинов разного строения различия в термодинамике рассматриваемых реакций незначительны.
Как показывает стехиометрия реакций, на их равновесие можно влиять, изменяя давление. Внутримолекулярной дегидратации, идущей с увеличением числа молей веществ, способствует пониженное или обычное давление. Наоборот, гидратации олефинов благоприятствует высокое давление, увеличивающее равновесную степень конверсии олефина. Так последняя при 250 – 300 *С и атмосферном давлении составляет всего 0,1 – 0,2%, что совершенно неприемлемо для практических целей, но при 7 – 8 МПа и тех же температурах она возрастает до 12 – 20%. Зависимость равновесной степени конверсии этилена при его гидратации от давления и температуры изображена на рис.60, причём аналогичные кривые характерны и для других олефинов. Очевидно, что гидратации способствуют одновременное снижение температуры и повышение давления. Рассмотрим теперь равновесие в системе межмолекулярная дегидратация спиртов – гидролиз простых эфиров. Термодинамическим методом регулирования направления этих реакций является изменение давления: на образование простого эфира оно не влияет, но получению олефина его снижение благоприятствует. Механизм и кинетика реакций Все рассматриваемые реакции принадлежат к числу кислотно-каталитических процессов. Типичными катализаторами гидратации являются достаточно сильные протонные кислоты: фосфорная кислота на носителе, поливольфрамовая кислота, сульфокатиониты. Для дегидратации используют фосфорную кислоту на носителе, оксид алюминия, серную кислоту, фосфаты (например СаНРО4) и другие. Роль катализаторов при гидратации состоит в протонировании олефина через промежуточное образование π- и σ-комплексов, причём обратная реакция дегидратации идёт через те же стадии, но в противоположном направлении: СН2=СН2 + Н+ = СН3=СН2+ + Н2О = СН3=СН2ОН + Н+
При межмолекулярной дегидратации карбокатион не отщепляет протон, а взаимодействует с другой молекулой спирта: СН3=СН2+ + СН3=СН2ОН = [СН3=СН+2] 2 О+Н = [СН3=СН+2] 2 О + +Н Электрофильный механизм дегидратации олефинов определяет уже отмеченные выше направления присоединения по правилу Марковникова, а также изменение реакционной способности олефинов, чем больше замещённость тем выше реакционная способность. В соответствии с этим этен самый нереакционноспособный. Для разных условий и катализаторов отношение реакционной способности олефинов меняется, составляя, например, для 80% серной кислоты 16 000: 1 000: 500: 1 и увеличиваясь для менее сильных кислот. Это очень существенно для выбора условий гидратации, особенно температуры: последняя может быть более низкой (и более благоприятной для равновесия) для изобутена по сравнению с пропиленом и особенно с этиленом. Равновесие гидратации – дегидратации мало зависит от строения олефина и спирта, поэтому ряд реакционной способности олефинов к гидратации должен соответствовать аналогичному ряду спиртов по их способности к дегидратации: Третичный > вторичный > первичный. Эта способность особенно растёт у β-кето- и β-нитроспиртов, электроноакцепторные группы которых повышают кислотность атомов водорода, находящихся при соседнем с НО-группой углеродном атоме. Это нередко делает возможным некаталитическую, дегидратацию или даже катализ реакции основаниями: –СОСН2–СНОН– + НО-; - Н2О = –СОСН-–СНОН– = –СОСН=СН– НО- При гетерогенно-каталитической внутримолекулярной и межмолекулярной дегидратации в газовой фазе кинетика процесса описывается соответственно следующими уравнениями:
Они учитывают практическую необратимость внутримолекулярной дегидратации и тормозящие влияние спирта и воды, лучше адсорбирующихся на активных центрах катализатора. При гидратации олефинов вода всегда находится в избытке, поэтому тормозящим влиянием спирта можно пренебречь:
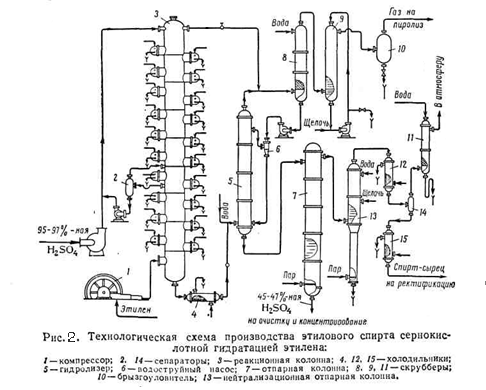
В ряде случаев роль воды более сложная. Так, фосфорная кислота, нанесённая на пористый носитель, образует на его поверхности жидкую плёнку, которая адсорбирует воду из газовой фазы. При каждых данных температуре и парциальном давлении водяных паров в газовой фазе устанавливается фазовое равновесие, и фосфорная кислота в плёнке имеет определённую концентрацию и соответствующую ей каталитическую активность. Последняя падает при снижении температуры и росте парциального давления воды, что ограничивает выбор этих параметров для каждого случая определёнными рамками. При катализе реакций гидратации – дегидратации при помощи сульфокатионитов было найдено такое кинетическое уравнение: Первый его член соответствует катализу сульфогруппами катионита, а второй – специфическому катализу ионами гидроксония Н3О+. если количество воды в смеси мало, в уравнении преобладает первое слагаемое, сильно зависящее от концентрации воды; повышение этой величины ведёт к преобладанию второго слагаемого. 4.2. Технология получения спиртов методом сернокислотной гидратации Схема установки получения этилового спирта сернокислотной гидратацией этилена приведена на (рис.2)
Исходным сырьём служит газообразная этан этиленовая фракция, содержащая 30 – 50 % этилена, и 95 – 97% серная кислота. Этиленсодержащий газ подают в реактор – абсорбционную колонну 3, орошаемую серной кислотой и имеющую 25 тарелок. Газ в виде мелких пузырьков барботирует на тарелках через слой жидкости, где происходит поглощение этилена. Для отвода тепла реакции на каждой тарелке имеется змеевик водяного охлаждения. В колонне поддерживается температура 65 – 75 °C и давление 25 кгс/см2 . Газ, покидающий колонну содержит 2 – 6 % этилена. Он проходит скрубберы 8 и 9 орошаемые соответственно водой и 5 – 10% раствором едкого натра, для отмывки серной кислоты и нейтрализации. Отмытый газ (этан) направляют на пиролиз. Из нижней части колонны 3 непрерывно вытекает реакционная масса, имеющая примерно следующий состав (%): этилсульфат – 60-80, серная кислота – 10-30, диэтилсульфат – 2-8, полимеры – 1-7, вода и прочие примеси -2. Эта смесь охлаждается в холодильнике 4 и направляется в гидролизёр 5, куда поступает и вода. Гидролиз проводится при 92 - 96°C и давлении 4,5 - 5 кгс/см2. из нижней части гидролизёра 5 реакционная масса поступает в верхнюю часть отпарной колонны 7, в низ которой подаётся острый пар. В нижней части колонны поддерживается температура 125 °C, в верхней 110°C и давление 0,5 кгс/см2. Из нижней части колонны 7 отводится 45 – 47 % серная кислота, поступающая на упаривание до 90 % концентрации. Пары спирта и других продуктов направляются в колонну 13, где отгоняется спирт – сырец. В эту колонну для нейтрализации серной кислоты вводят 5% раствор едкого натра. Спирт – сырец содержит 25 – 35% этанола, 3 – 5% диэтилового эфира, 60 – 65% воды и 0,05% полимеров. Его направляют на ректификацию для получения 95 - 96% концентрации спирта. Выход этанола ректификата составляет 85% от стехиометрического. Кроме того, получается диэтиловый эфир с выходом до 7%. Сернокислотная гидратация олефинов – самый распространённый метод получения спиртов. Однако недостатком метода является участие больших количеств серной кислоты и её разбавление, а отсюда необходимость её упаривания, перекачки больших объёмов и так далее. Всё это связано с коррозией аппаратуры и большими капитальными затратами на сооружение заводов. [2,5] 5. ПРОИЗВОДСТВО СПИРТОВ ПРЯМОЙ ГИДРАТАЦИЕЙ ОЛЕФИНОВ 5.1. Теоретические сведения В промышленности методом прямой гидратации получают этиловый и изопропиловый спирты. Прямая гидратация олефинов заключается в непосредственном присоединении воды к олефином: С2Н4 + H2O ↔ C2H5OH Синтез этилового спирта удалось осуществить лишь после того, как были изысканы достаточно активные катализаторы процесса. При газофазной гидратации в качестве катализаторов применяются фосфорная кислота или окись вольфрама на носителях. На последнем катализаторе процесс проводят и в жидкой фазе. Газофазная реакция прямой гидратации олефинов обратима и идёт с выделением тепла. Тепловой эффект зависит от строения исходных олефинов и их молекулярного веса: С2Н4 + H2O ↔ C2H5OH + 10,9 ккал/моль (45,6 кДж/моль);
Поскольку реакция идёт с выделением тепла и уменьшением объёма, ей благоприятствуют пониженные температуры и повышенные давления. Константа равновесия реакции равна: lgKр = (2100/T) – 6,195, где Т – температура, К. Практический выбор условий связан со скоростью реакции и, следовательно, с активностью катализатора. Реакцию удаётся реализовать при температурах от 200 до 300 °C, но эти условия термодинамически неблагоприятны для этилена. Поэтому на промышленных катализаторах степень конверсии олефинов в спирт низка. Также, как и в случае сернокислотной гидратации, присоединение воды происходит по правилу Марковникова. Механизм прямой гидратации олефинов в присутствии фосфорной кислоты был предложен Н.М.Чирковым. Первая стадия заключается в физическом растворении этилена в плёнке фосфорной кислоты на поверхности носителя. Затем происходит отщепление протона от молекулы кислоты: H3PO4 ↔ Н+ + H2PO4-
Известно, что олефины, как и ароматические углеводороды являются слабыми основаниями, поэтому прямую гидратацию олефинов можно рассматривать как реакцию электрофильного замещения. Этилен образует с протоном π-комплекс, который переходит в более стабильный ион карбония. Далее ион карбония взаимодействует с водой за счёт неподелённой электронной пары атома кислорода; в данном случае проявляется нуклеофильность воды, обладающей амфотерными свойствами. В результате образуется ион алкоксония, который отщепляет протон с образованием спирта: С2Н4 + Н+↔π-комплекс↔CH2+CH3 + H2O↔CH2(H2O) +CH3 + Н+↔C2H5OH В производстве этилового спирта прямой гидратацией этилена наиболее широкое применение получил фосфорнокислотный катализатор на твёрдом носителе. Катализаторы прямой гидратации не должны разрушаться под действием влаги, поэтому такой катализатор, как фосфорная кислота на кизельгуре, не применим – он не имеет скелета и легко разрушается. В качестве носителя для фосфорной кислоты применяют силикагель и алюмосиликат. Обычный шариковый алюмосиликат обрабатывают 20% -ной серной кислотой; при этом содержание оксида алюминия в нём снижается, а содержание оксида кремния повышается (излишнее количество оксида алюминия приводит к образованию малоактивных фосфатов алюминия). Затем носитель пропитывают 65%-ной фосфорной кислотой и сушат при 100°C. Готовый катализатор содержит 35-40% фосфорной кислоты. Если (как это и принято чаще всего) в качестве носителя используют шариковый силикагель, его обрабатывают водяным паром с целью пассивации. В условиях реакции фосфорная кислота, осаждённая на носителе, растворена в плёнке воды, адсорбированной на поверхности пор, и реакция фактически протекает в жидкой плёнке фосфорной кислоты. Кислотный катализ, таким образом, сводится к гомогенному катализу в жидкой плёнке катализатора. Как и в случае сернокислотной гидратации, при прямой гидратации этилена протекает ряд других реакций приводящих к побочным продуктам. За счёт взаимодействия иона карбония со спиртом образуется диэтиловый эфир: C2H5OH + CH2 +CH3 За счёт дегидрирования спирта образуется ацетальдегид C2H5OH
Причём реакция сопровождается образованием этана. Путём полимеризации этилена образуются полимеры: CH2+CH3
+ CH2CH2 По топытным данным конвертированный этилен расходуется на образование различных продуктов таким образом (в масс.%): на этанол – 94,5; на диэтиловый эфир – 2,5; на ацетальдегид – 2,0; на полимеры и сложные эфиры – 1,0. Реакция прямой гидратации этилена описывается следующим интегральным уравнением при Т, рH2O, рC2H4 – const:
Влияние температуры Реакцию прямой гидратации олефинов желательно проводить при невысоких температурах. Однако практически выбор температуры лимитируется скоростью реакции и активностью применяемых катализаторов.
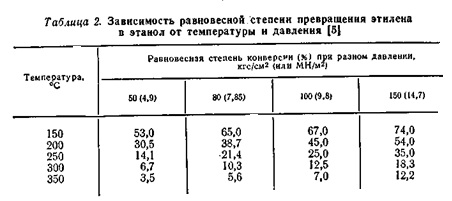
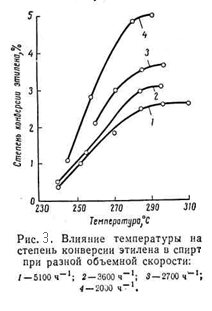
В таблице 2 приведена зависимость равновесной степени превращения этилена в этанол от температуры при разных давлениях. Из приведённых данных видно, что с повышением температуры равновесная стиепень превращения этилена в спирт снижается. Однако, при низких температурах активность фосфорнокислотного катализатора мала. Так, степень конверсии этилена при 280 – 290 °C достигает лишь 4 – 5%, а при более низких температурах она ещё меньше (рис.3.)
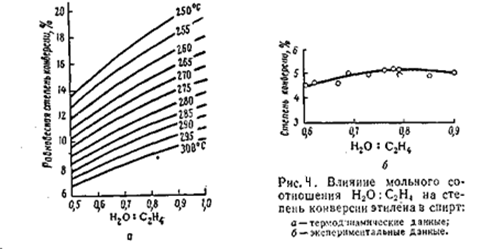
На практике процесс прямой гидратации в паровой фазе в присутствии фосфорнокислотного катализатора ведут в интервале 260 - 300°C. Влияние мольного соотношения воды и этилена Согласно термодинамическим данным, с увеличением отношения вода/этилен с 0,5/1 до 1/1 значительно повышается степень конверсии олефина
Однако экспериментальные данные отличаются от термодинамических (рис) с увеличением отношения вода/этилен до 0,7/0,75 степень конверсии этилена действительно возрастает, но при дальнейшем его увеличении она снижается. Установлено также, что от соотношения вода/этилен зависит и активность катализатора гидратации. Оптимальная концентрация фосфорной кислоты в жидкостной плёнке на пористом носителе составляет 83 – 85%. Эта величина зависит от парциального давления водяного пара, которое определяется общим давлением в системе и мольным отношением вода/этилен. Оптимальная концентрация фосфорной кислоты наблюдается при отношении вода/этилен = 0,75/1. с дальнейшим ростом этого отношения возрастает количество воды в плёнке, уменьшается концентрация кислоты и снижается степень конверсии этилена. Поэтому в промышленных условиях принято мольное отношение вода/этилен = (0,6 – 0,7)/1. Влияние давления Повышение давления благоприятствует реакции гидратации, причём оптимальное давление составляет 70 – 80 кгс/см2. это давление связано с процессом абсорбции этилена фосфорной кислотой. Оптимальное парциальное давление водяных паров равно 27 – 30 кгс/см2; оно и определяет мольное соотношение водяных паров и этилена. Оптимальное парциальное давление этилена составляет 36 – 38 кгс/см2.
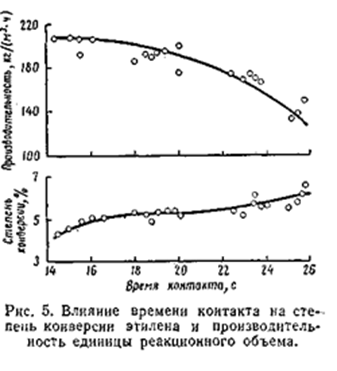
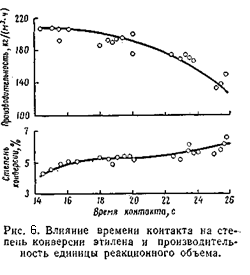
Общее давление складывается из парциальных давлениях воды, этилена и примесей. Прои концентрации этилена в циркулирующем газе 80 – 85% общее давление системы получается равным 70 - 80 кгс/см2. Влияние времени контакта С увеличением времени контакта степень конверсии этилена возрастает, а производительность единицы объёма снижается. Оптимальное время контакта 17 – 19 с, что соответствует объёмной скорости 1 800 – 2 000 (1/час). В оптимальных условиях степень конверсии этилена не превышает 4,2 – 4,5 %, а селективность его использования достигает 95%. [2,3]
5.2. Технологические особенности процесса Основной особенностью процесса прямой гидратации этилена является малая степень конверсии этилена за один проход – не выше 4,5%. Этим обусловлена необходимость рециркуляции значительных его количеств. Ввиду высокой кратности циркуляции этилена, в системе возможно накопление инертных примесей, поэтому содержание их в исходном этилене не должно превышать 2 – 5%. Эти примеси представляют собой метан и этан. В результате циркуляции непревращённого этилена концентрация примесей в циркулирующем этилене возрастает, а концентрация этилена снижается. Заданную концентрацию этилена в циркуляционном газе поддерживают путём отдувки части циркулирующего газа в систему газофракционирования. Поскольку в циркулирующем этилене инертных примесей больше, чем в свежем, при отдувке можно вывести из системы все поступающие туда примеси. Большие объёмы циркулирующего газа нужно охлаждать после реакции и вновь нагревать перед подачей в реактор, поэтому при гидратациит большую роль играет выбор эффективных способов охлаждения, подогрева и парообразования. Важное значение в процесс имеет также регенерация тепла, необходимая для снижения расхода пара или топлива на нагрев сырья и уменьшение расхода воды на охлаждение продуктов. Кроме того при рациональной схеме регенерации тепла может быть значительно понижен или полностью исключён расход пара высокого давления, необходимо для проведения собственной гидратации. Реакция прямой гидратации этилена идёт с выделением значительного количества тепла. Однако вследствие низкой конверсии этилена выделяющееся тепло расходуется на нагревание самого этилена и водяного пара, причём в реакторе адиабатического типа перепад температуры паро-газовой смеси не превышает 18 - 20°C, что вполне допустимо. Поэтому проблема отвода тепла в этом процессе не возникает. Ещё одной особенностью процесса является унос фосфорной кислоты вследствие пропускания значительного количества паро-газовой смеси через слой катализатогра. Унос кислоты пара-газовой смесью составляет 0,5 г/ч с 1л катализатора или 1,5 – 3 кг в расчёте на 1т спирта. Активность катализатора в процессе работы снижается вследствие уноса теплоты и зауглероживания. Срок службы катализатора составляет 400 – 500 ч. Затем катализатор регенерируют путём выжигания кокса и нанесения фосфорной кислоты. Срок службы катализатора можно увеличить до 900 – 1000 ч, добавляя фосфорную кислоту в парогазовую смесь на входе в реактор. В качестве сырья для процесса прямой гидратации используется технический этилен, содержащий 95 – 98% этилена. 5.3. Технологическая схема процесса Процесс прямой гидратации этилена включает следующие стадии: 1) компримирование свежего этилена и дополнительное компримирование циркулирующего этилена; 2) нагревание этилена, приготовление паро-газовой смеси и её подогревание; 3) контактирование парогазовой смеси с катализатором; 4) нейтрализацию фосфорной кислоты, уносимой из реактора; 5) охлаждение парогазовой смеси и конденсацию паров спирта и воды; 6) ректификацию спирта – сырца.
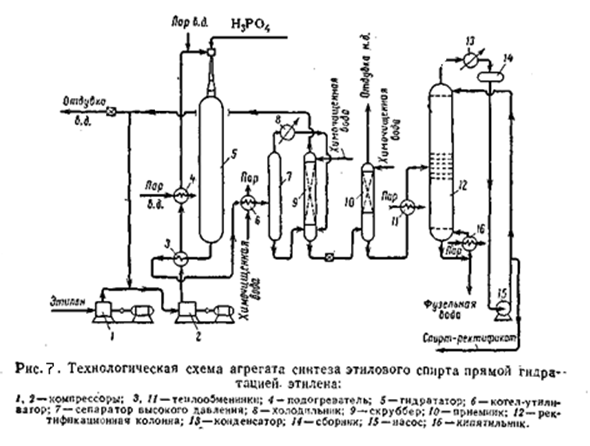
На рисунке приведена принципиальная технологическая схема агрегата синтеза этанола прямой гидратацией этилена. Свежий этилен под давлением 20 – 23 кгс/см2 поступает на приём компрессора 1, сжимается до 70 кгс/см2 и смешивается с циркулирующим этиленом. Газовая смесь циркуляционным компрессором 2 сжимается до 80 кгс/см2 и направляется в теплообменник 3, где подогревается за счёт тепла обратного газа.
Далее газ нагревается в подогревателе 4 паром высокого давления до 220°C. Такая температура установлена для начала цикла синтеза. В конце цикла эта температура должна достигать 260°C. После подогревателя 4 прямой газ смешивается с перегретым (450°C) паром высокого давления (80 кгс/см2); в результате температура смеси составляет 275°C в начале цикла и 285 - 290°C в конце. В смесителе, установленном в верхней части гидрататора 5, паро-газовая смесь смешивается с 7% фосфорной кислотой и поступает в гидрататор 5. Газ проходит слой катализатора сверху вниз, причём за счёт реакционного тепла температура повышается на 18 – 20 °C, то есть до 293 °C в начале цикла и до 303 – 308 °C в конце. Выходящий из гидрататора реакционный газ уносит с собой некоторое количество фосфорной кислоты. Нейтрализация её осуществляется впрыскиванием в реакционный газ щелочного спирта-водного конденсата. Температура газа при этом снижается до 220 °C. После нейтрализации реакционный газ проходит теплообменник 3, где охлаждается с 220 до 194°C, и далее котёл-утилизатор 6, где генерируется пар давлением 5 кгс/см2. Из котла утилизатора газ и сконденсировавшаяся жидкость (145°C) направляется в сепаратор 7. отделённый от жидкости газ из сепаратора охлаждается в холодильнике 8, где конденсируются пары спирта и воды. Несконденсировавшийся газ и конденсат из холодильника 8 направляются в насадочный скруббер 9, где остатки этанола поглощаются умягченной водой. Газ из скруббера идёт на смешение со свежим этиленом. Для вывода из системы накопившихся инертных примесей часть газа через регулятор давления сбрасывают в цех компрессии. Спирто-водный конденсат из скруббера дросселируют и направляют в приёмник 10. выделившийся при дросселировании газ отмывают от паров спирта умягчённой водой в насадочной части приёмника 10 и через регулятор давления направляют в цех компрессии. Спирто-одный конденсат из приёмника 10 нагревают до 80°C в теплообменнике 11 и направляют на ректификацию в колонну 12. Температура на верху колонны 80°C, внизу 109°C; давление 1,1 – 1,4 кгс/см2. подвод тепла в низ колонны осуществляют через кипятильник 16, обогреваемый паром давлением 3 кгс/см2 из котла-утилизатора. Пары спирта (азиотропная смесь) с верха колонны 12 поступают в конденсатор 13, где конденсируются и охлаждаются до 75 °C. Конденсат стекает в сборник 14, откуда часть спирта подаётся насосом 15 на орошение колонны 12. Остальное количество выводится с установки. Фузельная вода с низа колонны выводится в канализацию. Рассмотренная схема обладает рядом недостатков; в первую очередь, велик расход водяного пара высокого давления. Кроме того, унос фосфорной кислоты парогазовой смесью приводит к необходимости нейтрализации смеси путём впрыскивания щелочного раствора спирто-водного конденсата; это снижает температуру паро-газовой смеси и уменьшает возможности регенерации тепла. Использование пара высокого давления можно полностью исключить за счёт генерации пара в системе теплообмена. Для этого в поток прямого газа подают химически очищенную воду под давлением, и в процессе теплообмена с обратным газом вода испаряется. Теплообмен осуществляется в специальных теплообменниках сатураторах.
При этом степень насыщения газа парами воды достигает 0,6 – 0,7 моль/моль, а конечная температура парогазовой смеси равна 215°C. Подогревание парогазовой смеси до 275°C осуществляется в трубчатой печи за счёт сжигания топлива, таким образом, дополнительный расход пара высокого давления исключается. Изготовление теплообменника 2 из омеднённых труб, а трубные решётки из биметалла сталь-медь позволяет исключить нейтрализацию парогазовой смеси и интенсифицировать регенерацию тепла обратного газа. Технологическая схема усовершенствованного агрегата прямой гидратации этилена приводится на рисунке
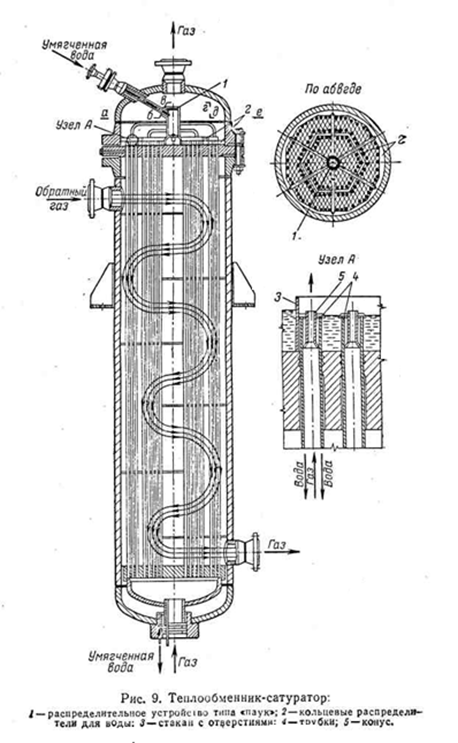
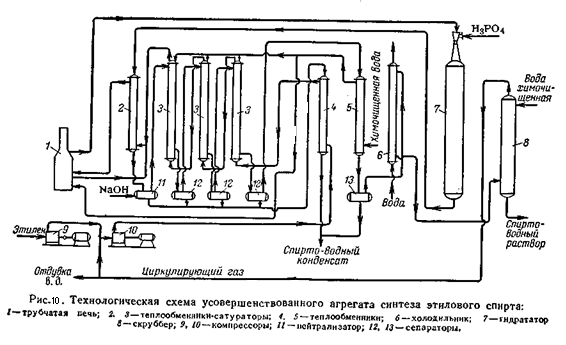
Компримированный этилен после компрессоров 9 и 10 поступают в теплообменник 4, где нагревается до 100 °C конденсатом после теплообменников-сатураторов (180°C). Далее этилен последовательно проходит теплообменники-сатураторы 3, орошаемые конденсатом. Насыщенный водой этилен при 200°C поступает в омеднённый теплообменник-сатуратор 2, а перед входом смешивается с частично испариной химически очищенной водой из теплообменника 5. Смесь при 215°C поступает в трубчатую печь 1, там догревается до 270 - 290°C и направляется в гидрататор 7, работающий в интервале 260 – 300°C. Обратный газ из гидратора проходит омеднённый теплообменник-сатуратор 2, где охлаждается до 240°C, и затем нейтрализуется щелочным раствором спирто-водного конденсата в нейтрализаторе 11; при этом температура снижается до 220 °C. Далее обратный газ последовательно проходит все теплообменники-сатураторы 3 и отделяется от жидкости в сепараторах 12, после чего при 130°C направляется на окончательное охлаждение в теплообменник 5 и холодильник 6 и поступает в скруббер 8 для отмывки спирта. Спирто-водный конденсат из сепараторов поступает в теплообменник 4. Все потоки спирто-водного конденсата и спирто-водный раствор после освобождения от растворённого этилена поступает затем на ректификацию.[2,4] 5.4. Характеристика основной аппаратуры Реактор (гидрататор) представляет собой пустотелый цельнокованый цилиндрический стальной аппарат внутренним диаметром 1260 – 2200 мм и толщиной стенки 70 мм, футерованный слоем меди толщиной 12 – 15 мм. (рис а)
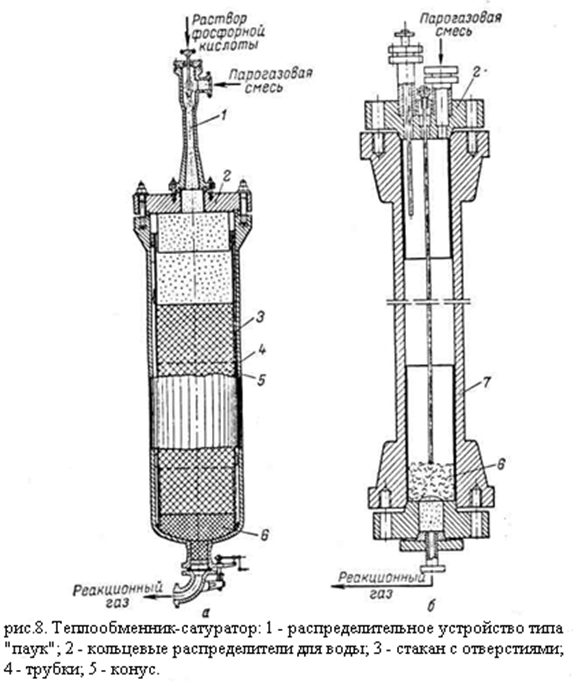
В качестве усовершенствования реактор может быть изготовлен из биметалла сталь-медь (рис б). Высота слоя катализатора приблизительно 7 метров. Кроме того, для снижения уноса кислоты в нижнюю часть реактора загружают слой чистого носителя высотой 1 метр. Линейная скорость газа 0,2 м/с; потеря напора 3 – 4 кгс/см2 в начале цикла работы и до 6 кгс/см2 в конце. Теплообменник-сатуратор - вертикальный кожухотрубный аппарат, в котором прямой газ проходит по трубам, а обратный – по межтрубному пространству; прямой газ поступает снизу вверх. Вода подаётся сверху, распределяется по трубной решётке через специальное устройство – «паук», растекается по решётке и поступает в трубы через кольцевые зазоры между трубками и вставленными в их верхнюю часть конусами. Благодаря этому вода стекает тонкой плёнкой по поверхности трубок испаряясь и насыщая поднимающийся навстречу прямой газ. Насыщенный водой прямой газ через конусы попадает в пространство над трубной решёткой. Коэффициент теплопередачи в теплообменниках-сатураторах достигает 400 ккал/(м2 . ч . °C).[2,5] 5.5. Расчёт материального баланса гидратора При прямой гидратации этилена на фосфорнокислотных катализаторах помимо основного процесса получения этанола из этилена, протекают побочные реакции: 1)образование диэтилового эфира; 2)образование ацетальдегида; 3)образование полимеров. Задаёмся количеством образующегося спирта Gc (кг/ч). Обозначим доли конвертируемого этилена, расходуемого на образование различных продуктов (в масс.%): на этанол – С1; на диэтиловый эфир – С2; на ацетальдегид и этан – С3; на полимеры – С4. Расход этилена Расход этилена рассчитывают, исходя из заданного распределения вступившего в реакцию этилена и стехиометрического уравнения реакции. Общий расход этилена равен:
где Мэ и Мс – молекулярные веса этилена и спирта. Из этого количества расходуются: на образование этанола
На образование диэтилового эфира:
На образование ацетальдегида и этана:
На образование полимеров:
Количество продуктов реакции Количество этилового спирта заданно Gc (кг/ч). Количество побочных продуктов (кг/ч) находим на основе стехиометрических уравнений. Количество диэтилового эфира равно:
Количество ацетальдегида равно:
Количество этана равно:
где Мэф и Мэт – молекулярные веса эфира и этана. Рассчитываем количество полимеров. Условно считаем, что полимеры состоят только из углерода и водорода. Тогда имеем: Gп = А4, кг/ч. Количество воды на реакцию. На каждую из реакций расходуется вода в количестве 1 моль на 1 моль продукта. Общее количество воды составляет:
Количество отдуваемого газа. Для поддержания определённой концентрации этилена в циркулируемом газе необходимо выводить из системы поступающие инертные примеси путём отдувки части циркуляционного газа. Инертные примеси поступают в систему с исходным техническим этиленом, а также за счёт побочного образования небольших количеств этана. Хотя общее количество этих примесей невелико, они неизбежно будут накапливаться в системе, если их не удалять.
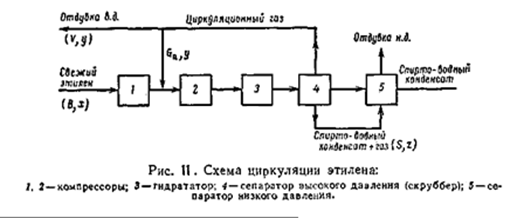
На рисунке приведена принципиальная схема циркуляции этилена. В гидратор 3 поступает компримированный этилен. Парогазовая смесь из гидратора после конденсации разделяется на газ и конденсат в сепараторе 4 высокого давления поскольку этот аппарат работает под давлением часть газа остаётся растворённой в спирто-водном конденсате. Последний после дросселирования направляется в сепаратор 5 низкого давления, где отделяется растворенный газ – отдувка низкого давления (н.д.). циркуляционный газ высокого давления из сепаратора 4 поступает на приём компрессора 2, а часть газа отдувается в систему газофракционирования (отдувка в.д.). таким образом, инертные примеси удаляются из примеси двумя путями – с отдуваемым газом высокого давления и с растворённым газом сепаратора 4 (отдувка н.д.). вместе с отдуваемым и растворённым газом из системы выводится также этилен. Поэтому количество свежего этилена, который необходимо подать в систему превышает количество конвертируемого этилена и зависит от количества отдуваемого этилена. Введём следующие обозначения: А – количество конвертируемого этилена, кг/ч или кмоль/ч; В – количество свежего технического этилена, кг/ч или кмоль/ч; V – количество отдуваемого газа высокого давления, кг/ч или кмоль/ч; S – количество газа растворённого в спирто-водном конденсате, кг/ч или кмоль/ч; Gэт – количество образующегося этана, кг/ч или кмоль/ч; x, y, z – массовые или мольные концентрации этилена соответственно в свежем техническом этилене, в циркуляционном газе и в растворённом газе. Составим баланс по этилену. Количество этилена, поступающего в систему, равно сумме конвертированного, отдуваемого и растворённого этилена Bx = A + Vy + Sz (1) Точно также составим баланс по примесям. Они поступают в систему со свежим этиленом плюс образующийся по реакции этан; примеси выводятся из системы с отдувкой в.д. и с растворённым газом. Следовательно, можно записать равенство: B(1– x) + Gэт = V(1– y) + S(1 – z) (2) Количество свежего этилена В неизвестно. Найдём его значение из уравнения (1): В = (А + Vy +Sz)/ x (3) И подставим его в уравнение (2):
Преобразуем уравнение (4): (A + Vy + Sz)(1 – x ) + Gэт.x = V(1 – y)x + S(1 – z)x; A(1 – x) + Gэт.x = V(x – y) + S(x – z); V(x – y) = A(1 - x) + Gэт.x – S(x – z). Количество отдуваемого циркуляционного газа выразится уравнением:
Из уравнения (5) следует, что V увеличивается с уменьшением концентрации свежего этилена (х) и с увеличением концентрации этилена в циркуляционном газе (у), а из уравнения (1) следует, что с увеличением V увеличивается количество свежего этилена, которое необходимо подать в систему. Поэтому практически подбирают такие концентрации х и у, которые обеспечивали бы оптимальное парциальное давление в системе при приемлемом с экономической точки зрения количестве отдуваемого газа высокого давления. Количество отдуваемого чистого этилена равно: Vэ = Vy, кг/ч или кмоль/ч; Количество отдуваемых примесей составляет: Vпр = V(1 - y) кг/ч или кмоль/ч; Концентрацию этилена в циркуляционном газе находим в материальном баланса по метану и этану:
В.хметан = V.yметан +S.zметан (6) В.хэтан + Gэт = V.yэтан +S.zэтан (7) Значение В легко вычислить из уравнения (2); следовательно, оно уже известно, поэтому из уравнения (6) и (7) легко найти соответствующие концентрации: yметан = (В.хметан – S.zметан )/V (8) yэтан = (В.хэтан + Gэт – S.zэтан )/V (9) Для расчёта отдуваемого газа и последующих расчётов задаются значениями х и у. значение S и Z, zметан и zэтан определяют при расчёте сепаратора высокого давления, так как они заранее не известны, ими задаются на основе опытных данных методом подбора. Количество прямого газа, поступающего в гидратор, определяют как сумму циркулирующего и свежего этилена. Зная количество каждого компонента, определяем состав прямого газа: метан – Gц. yметан + В.хметан (кг/ч), этилен – Gэпр; этан – Gц. yэтан + В.хэтан . .Всего - Gпр. Количество водяного пара, подаваемого в гидратор, равно: Nz = nz . Gэпр/28, кмоль/час. Gz= 18 Nz кг/ч Количество обратного газа, выходящего из гидратора составит: Gобр.в. = Gпр + Gz – A – GH2O + Gспирт + Gэф + Ga + Gэт + Gп, кг/ч. [1] 6. ПРЯМАЯ ГИДРАТАЦИЯ ЭТИЛЕНА НА НЕЙТРАЛЬНЫХ КАТАЛИЗАТОРАХ Существенными недостатками фосфорнокислотного катализатора являются его коррозионная агрессивность и постепенный унос кислоты с поверхности носителя. Эти недостатки могут быть устранены при использовании нейтральных катализаторов – вольфрамовых и кремний-вольфрамовых. Разработаны процессы гидратации этилена на нейтральном катализаторе в жидкой фазе при 250 - 300°C и 300 кгс/см2 и при 300 °C и 140 кгс/см2. в этих случаях процесс ведут в колонне высокого давления, где на тарелках помещён катализатор – окислы вольфрама на силикагеле. Этилен и воду подают в верхнюю часть колонны, а снизу отводят 20% спирт. При этом процессе не требуется расходовать большое количество тепла на испарение воды и перегрев водяного пара. Разработаны другие активные вольфрамовые катализаторы, содержащие 40 – 60% трёхокиси вольфрама на широкопористом силикагеле типа SiO2. 12WO3. 7H2O с добавкой борной кислоты. Катализаторы этого типа готовят, пропитывая силикагель раствором вольфромата аммония и прокаливая затем при 400°C. Они не нуждаются в последующем восстановлении. Добавление в состав катализатора 5 -10% борной кислоты существенно повышает его активность. Наиболее активный катализатор, содержащий 60% оксида вольфрама (VI) и 5% В2О3 на силикагеле, применяется в интервале 200 – 240 °C и 15 – 25 кгс/см2 , то есть в более мягких условиях, чем фосфорный катализатор. Сравнение работы этих катализаторов на пилотной установке приведено в таблице Содержание спирта в конденсате при гидратации этилена на кремневольфрамовых катализаторах составляет 12 – 13%.
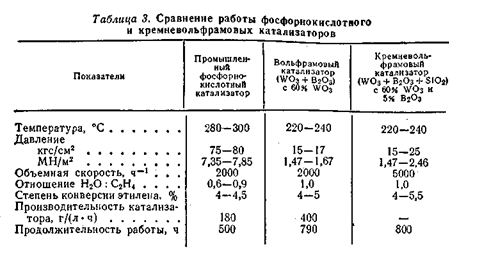
Вольфрамовые катализаторы помимо уже указанных преимуществ более активны, чем фосфорнокислотные и могут работать в более мягких условиях. Однако они значительно дороже, ибо вольфрам дефицитный материал.[2,6] ЗАКЛЮЧЕНИЕ Было рассмотрено три вида гидратации этилена: прямая гидратация с использованием в качестве катализатора фосфорной кислоты, сернокислотная гидратация этилена и гидратация на нейтральных катализаторах. Последняя, как уже было упомянуто выше, из-за своей дороговизны используется крайне редко. Большее распространение в промышленности получили первые два метода. В случае сернокислотной гидратации основной статьёй расхода является сырьё и вспомогательные материалы, что связано с применением серной кислоты и с меньшей селективностью процесса, протекающего с образованием значительных количеств побочных продуктов. Себестоимость спирта полученного сернокислотной гидратацией примерно на 20% выше, чем при прямой гидратацией. Поэтому с экономической точки зрения процесс прямой гидратации является наиболее выгодным. Уже было упомянуто, что при сернокислотной гидратации образуется множество побочных продуктов, к ним относятся: диэтилсульфат, этилсульфат, диэтиловый эфир, этаналь и различные продукты полимеризации этилена (олигомеры и полимеры). Кроме того, сама серная кислота является опасным продуктом. Все эти вещества в больших количествах оказывают вредное воздействие на окружающую среду. И поэтому их нужно утилизировать, что увеличивает затраты на производстве. При прямой гидратации тоже образуются побочные продукты: диэтиловый эфир, этаналь, фосфорные эфиры и различные виды полимеров, остатки фосфорной кислоты. Но в значительной меньшей мере, чем при сернокислотной гидратации этилена. Поэтому с точки зрения экологичности процесса метод прямой гидратации этилена более выгоден. [2, 7] СПИСОК ИСПОЛЬЗОВАНОЙ ЛИТЕРАТУРЫ 1. В.И.Коробкин, Л.В.Передельский. Экология. Ростов н/Д: издательство «Феникс», 2003. С. 300 – 302. 2. «Химическая энциклопедия», т.1, М.:Химия, 1987г – 566с. 3. «Химическая энциклопедия», т.2, М.:Химия, 1987г – 540с. 4. Несмеянов А. Н., Несмеянов Н.А. «Начала органической химии», т.2, М.:Химия, 1969 – 826 с. 5. Лебедев Н.Н. «Теория химических процессов основного органического и нефтехимического синтеза». М.:Химия, 1988 – 588 с. 6. Несмеянов А. Н., Несмеянов Н.А. «Начала органической химии», т.1, М.:Химия, 1969 – 668с. 7. Степанов А.В. «Производство этилена» Киев, издательствово «Наукова думка» 1973г. – 400с. |