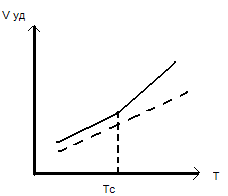
Курсовая работа: Стеклование и стеклообразное состояниеКурсовая работа: Стеклование и стеклообразное состояниеСТЕКЛОВАНИЕ И СТЕКЛООБРАЗНОЕ СОСТОЯНИЕ Температура, при которой полимер при охлаждении переходит из высокоэластического или вязкотекучего состояния в стеклообразное, называется температурой стеклования. Полимеры в стеклообразном состоянии отличаются рядом особенностей релаксационного поведения и комплекса механических свойств от полимеров в высокоэластическом состоянии. Это становится очевидным при сравнении, например, свойств натурального каучука (типичный эластомер) и полиметилметакрилата, часто в обиходе называемого органическим стеклом. СТЕКЛОВАНИЕ В высокоэластическом состоянии полимеры имеют в целом неупорядоченную надмолекулярную структуру, в которой имеются как более упорядоченные, так и менее упорядоченные элементы. Поэтому говорят, что полимер имеет жидкостную структуру, которая характеризуется наличием ближнего порядка. В полимере понятие ближнего порядка относится не к молекулам, а к их сегментам, которые образуют ассоциаты (узлы флуктуационной сетки) с наиболее выраженным ближним порядком. В низкомолекулярной жидкости регулярность в расположении молекул существует только между соседними молекулами; уже на расстоянии 4 или 5 молекулярных диаметров эта регулярность полностью исчезает. В полимерах размеры упорядоченных областей могут быть много больше. При охлаждении значительно уменьшается свободный объем. Коэффициент теплового расширения (сжатия) полимеров в эластическом состоянии составляет (6÷7) ∙10-4 1/град. Можно полагать, что при достаточном охлаждении свободный объем должен уменьшиться до нуля, но в действительности этого не происходит, поскольку сегменты макромолекул, в которые входит по 5— 20 атомов углерода, могут участвовать в тепловом движении лишь при наличии достаточных по размеру пустот (или дырок) по соседству с ними. В эти дырки и перемещаются сегменты в процессе теплового движения. Если свободный объем становится менее 2,5 % от общего объема полимера, размеры «дырок» и их число становятся настолько малыми, что тепловое перемещение сегментов прекращается. Таким образом, при охлаждении полимер перестает быть эластичным и становится твердым, стеклообразным уже тогда, когда свободный объем достигает 2,5 % от объема тела. Температура, при которой это происходит, и есть температура стеклования Тс. На рис. 1 показано уменьшение удельного объема при понижений, температуры. Видно, что после достижения Тс удельный объем при дальнейшем охлаждении меняется гораздо медленнее. Действительно, для полимеров в стеклообразном состоянии коэффициент теплового расширения составляет только 2∙10-4 1/град. В области стеклообразного состояния изменение ближнего порядка при охлаждении уже не происходит и удельный объем уменьшается только за счет уменьшения расстояний между молекулами. Расчет показал, что если бы полимер сохранял способность к изменению ближнего порядка при охлаждении до Т< Тс, то можно было бы достичь нулевого значения свободного объема при Т2 на 51,6 С ниже Тс. Теоретически достичь Т2, меньшей Тс на 51,6 оС, можно при бесконечно медленном охлаждении. Переход полимера в стеклообразное состояние при охлаждении носит название структурного стеклования. Это значит, что этот переход сопровождается фиксацией определенной структуры, определенного ближнего порядка, которые не меняются при дальнейшем охлаждении. Фиксация структуры, исключение возможности ее перестройки при охлаждении делают стеклообразный полимер неравновесным. Это в первую очередь приводит к зависимости Тс от скорости охлаждения. При медленном охлаждении сегменты успевают перемещаться даже при приближении к Тс и требуется сильно охладить полимер, чтобы предотвратить вся кие перестройки структуры. Излом на кривой зависимости удельного объема от Т (см. рис. 1) сместится в область более низких температур. Так, выдерживая образец поливинилацетата при каждой температуре в одном опыте в течение 0,02 ч, а в другом - 100 ч, получим значения Тс соответственно 32 и 23оС, т.е. отличающиеся на 9 о С.
Рис. 1. Зависимость удельного объема ( Vуд) от температуры. Температурное изменение свободного и занятого объемов Полимер становится малодеформируемым, как бы стеклообразным, не только при охлаждении до Тс, но к при более высокой постоянной температуре с уменьшением продолжительности действия силы или (что то же самое) с ростом скорости действия силы. Мы наблюдаем при этом механическое стеклование — потерю способности полимера к высокоэластической или вязкотекучей деформации при большой скорости действия силы. Структурное стеклование наступает при Т= Тс, механическое — при τ = t, т. е. когда критерий Деборы D = 1*. * Сказанное является условным: равновесная температура стеклования на 51,6 оС ниже практически определяемой, а механическое стеклование наступает при t <τ, т.е. при больших частотах, так как условие t =τ При механическом стекловании структура полимера не фиксируется, тепловое движение сегментов не прекращается. Однако скорость теплового движения оказывается меньше скорости действия силы, и заметные деформации не успевают развиваться: полимер ведет себя, как застеклованный. Чем больше скорость действия силы, тем выше Тс при механическом стекловании. Чем выше скорость охлаждения, тем выше Тс при структурном стекловании. Это значит, что стеклование есть не структурный (фазовый), а релаксационный переход, определяемый не перестройкой надмолекулярной структуры, а величиной отклика системы на внешнее воздействие. Это отличает стеклование от фазовых переходов, таких, например, как кристаллизация или плавление, при которых происходит качественное изменение структуры. При кристаллизации выделяется теплота кристаллизации, при стекловании тепловой эффект отсутствует. При кристаллизации скачкообразно уменьшается свободный объем; при стекловании объем не меняется, а излом на кривой Vуд—Т обусловлен лишь разными коэффициентами теплового расширения в эластическом и стеклообразном состояниях (рис. 1). Имеются и другие отличия, указывающие на то, что стеклование является релаксационным переходом, а не фазовым переходом первого или второго рода. Существует довольно много способов определения Тс. Их можно приблизительно разделить на две группы. Первая группа включает в себя методы, связанные с определением свойств полимера, температурная зависимость которых различна в эластическом и стеклообразном состояниях. Вторая группа включает в себя методы непосредственного определения изменения подвижности сегментов с температурой. В момент расстеклования образца (перехода в высокоэластическое состояние) наблюдается резкий рост молекулярной подвижности. соответствует середине интервала переходной области термомеханической кривой (точка перегиба на кривой модуль—частота). Рассмотрим первую группы методов определение Тс Температурная зависимость удельного объема. Точка перегиба прямой (см. рис. 1) определяет Тс. Удельный объем определяют обычными способами, например дилатометрически. Зависимость теплоемкости от температуры. В точке стеклования наблюдается скачок теплоемкости. Современные сканирующие приборы позволяют вести нагревание тонких образцов со скоростью до десятков градусов в минуту (дифференциальная сканирующая калориметрия). При этом измеряют чаще всего не теплоемкость, а температуру в образце, который нагревается с постоянной скоростью. При переходе из стеклообразного состояния в высокоэластическое теплоемкость резко увеличивается, что при водит к временному падению температуры, которая затем снова растет при дальнейшем нагревании образца. Минимум температуры образца определяет положение Тс. Показатель преломления. Зависимость показателя преломления от температуры выражается прямой с изломом в точке стеклования. Термомеханические кривые. По кривой, полученной в координатах деформация ε (модуль G)—температура, находят температуру механического стеклования, которая зависит от времени действия силы. Так, Тс натурального каучука равна -56 оС при частоте действия силы ω= 0,167 с-1 и —14 оС при ω = 2 ∙ 106 с-1 . Установлено однако, что если время действия силы не выходит за пределы от нескольких секунд до десятков минут, то значение Тс практически совпадает с температурой структурного стеклования. Учитывая, что точность определения температуры стеклования обычно не более 0,5—1,0 оС, временные интервалы действия силы можно еще более расширить без заметного изменения значения Тс. Термомеханический метод определения Тс наиболее широко распространен благодаря его простоте. Определяют зависимость оттемпературы разных механических показателей, таких как модуль, де формация, твердость, податливость, тангенс угла механических потерь. Последний особенно предпочтителен, поскольку зависимость tg δ— Т выражается кривой с максимумом, по положению которого можно более точно определить Тс, чем по другим терме механическим кривым, на которых в точке стеклования наблюдается перегиб. Во второй группе методов определения Тс рассмотрим три наиболее важных. Диэлектрический метод. При помещении образца полярного полимера в электромагнитное поле диполи полимера начинают следовать за изменением поля как раз в момент расстеклования что приводит к появлению максимума на кривой зависимости тангенса диэлектрических потерь от температуры. Зависимость Тс от частоты поля здесь существует, как в термомеханическом методе. Метод радиотермолюминесценции (метод РТЛ). Застеклованный образец подвергают ионизирующему облучению (например, гамма-облучению). Часть электронов, оторвавшихся при этом от полимера, попадает в «ловушки», в качестве которых выступают раз личные дефекты структуры, места концентрации свободного объема, примеси и т. п. В момент расстеклования сегменты становятся подвижными, электроны освобождаются из ловушек и взаимодействуют с ионизированным полимером. Выделяющаяся энергия вызывает свечение, которое можно зафиксировать чувствительными приборами. После исчерпания всех электронов излучение прекращается. Наблюдаемый пик радиотермолюминесценции определяет значение Тс на температурной шкале. Метод ядерного магнитного резонанса. Метод ЯMP позволяет определить подвижность ядер водорода. Поскольку последние связаны с сегментами полимера, увеличение подвижности в системе с ростом температуры позволяет определить Тс. ЗАВИСИМОСТЬ Тс ОТ СТРУКТУРЫ ПОЛИМЕРА
Зависимость от молекулярной массы. При малых степенях полимеризации (область олигомеров) Тс растет с ростом молекулярной массы. Это объясняется ростом массы кинетической единицы, перемещающейся как единое целое (это молекула олигомера). Если молекулярная масса достигает размеров, обеспечивающих ее гибкость, то в элементарном акте теплового движения перемещается сегмент, а не молекула. Размер сегмента не зависит от молекулярной массы макромолекулы (растет только число сегментов), поэтому и Тс остается постоянной (стремится к пределу, характерному для высокомолекулярного полимера). Это видно из рис. 2, где зависимость Тс—М приведена для полистирола. У гибкоцепных полимеров, таких как полиизобутилен, Тс достигает предела при М = 1000, а у более полярных полимеров Тс = const при М= (12÷40)∙103 и более. Влияние полярности полимера. С увеличением полярности растет межмолекулярное взаимодействие и величина энергетического барьера на пути перескока сегмента. Увеличивается температура, при которой становится возможен перескок, т. е. повышается Тс. Так, в ряду бутадиен-нитрильных сополимеров Тс повышается с ростом содержания нитрильных групп: Полимер... СКН-18 СКН-26
СКН-40 ПП ПС
Рис 2. Зависимость температуры стеклования от молекулярной массы фракций полистирола.
Влияние размера боковых заместителей. С ростом размера заместителей увеличивается масса сегмента, часто параллельно возрастает полярность и повышается Тс: Полимер............ПЭ ПП ПС Поли-2,6-дихлорстирол Тс, оС.................-65 -10 +100 +167 В то же время противоположный эффект наблюдается, когда основная цепь полимера, как, например, у ПММА, полярна, а заместители неполярны. Тогда неполярные заместители снижают общий уровень межмолекулярного взаимодействия (уменьшается энергия когезии), и понижается Тс с ростом длины неполярного заместителя:Полимер..Тс Полиметилметакрилат............................110 Полиэтил метакрилат..............................65 Поли-н-бутилметакрилат........................21 Поли-н-октилметакрилат.......................—20 Симметрично расположенные заместители компенсируют заряды полярных групп, энергия когезии уменьшается, и снижается Тс. Так, для ПП Тс = -10 °С, а для ПИБ Тс = -70 °С; для ПВХ Тс = 85 оC, а для поливинилиденхлорида Тс = 19 °С.
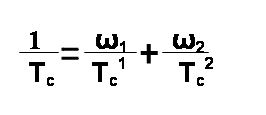
Где Тс1 и Тс2 — температуры стеклования соответствующих гомополимеров, а ω2 — массовые доли мономера в макромолекуле сополимера. Уравнение Флори—Фокса дает кривую, слегка отклоняющуюся от аддитивной (прямолинейной) зависимости в сторону оси составов. Это видно из следующих значений Тс сополимеров бутадиена и стирола: Полимер ПС СКС-90 СКС-80 СКС-60 СКС-10 Тс ,о С ...100 +84 +14 -24 -80 Влияние давления. Рост гидростатического давления приводит к объемному сжатию. При этом уменьшается свободный объем и растет Тс. Влияние пространственных сшивок. Возникновение поперечных связей в полимере приводит к его уплотнению, т. е. к уменьшению свободного объема и росту энергии когезии. Это, в свою очередь, приводит к повышению Тс с увеличением густоты сшивания приблизительно в соответствии с уравнением
Чем меньше Мс (больше густота сшивания), тем сильнее растет Тс. Приведенная формула больше применима к каучукам, а для пластмасс числовые коэффициенты другие. Влияние пластификации. Пластификаторы понижают Тс и тем самым расширяют интервал, в котором полимер сохраняет гибкость и деформируемость. Пластификаторы — это жидкости, обладающие малой летучестью, высокой температурой кипения и низкой температурой замерзания. В них свободный объем больше, чем свободный объем полимера, поэтому введение пластификатора увеличивает свободный объем в системе и приводит к снижению Тс. Увеличение свободного объема приводит также к снижению межмолекулярного взаимодействия. Снижение межмолекулярного взаимодействия особенно выражено, если в полимере есть сильные межмолекулярные связи, например водородные. Если пластификатор подобран правильно, он экранирует полярные группы, препятствуя образованию связей полимер—полимер. Это также снижает Тс. Чем больше добавлено пластификатора, тем сильнее снижается Тс. Неполярный пластификатор понижает Тс неполярного полимера (∆Тс) пропорционально объемной доле пластификатора (φ) в полимере (эффект разбавления): ∆Тс =k' φ Полярный пластификатор понижает Тс полярного полимера пропорционально мольной доле пластификатора (N) в полимере (экранирование полярных групп): ∆Тс = k''N МЕХАНИЧЕСКИЕ СВОЙСТВА ПОЛИМЕРНЫХ СТЕКОЛ Поскольку в стеклообразном состоянии сегменты лишены возможности совершать тепловые перемещения в соседние положены из-за отсутствия достаточного свободного объема, можно сделать вывод, что стеклообразный полимер не способен к большим деформациям. В действительности же стеклообразный полимер способен деформироваться без разрушения на сотни процентов, хотя и не способен самопроизвольно сокращаться после снятия нагрузки. На рис. 3 приведена кривая напряжение—деформация стеклообразного полимера. Весь процесс растяжения условно делится на три стадии. На первой стадии полимер растягивается упруго. Деформация достигается за счет увеличения межмолекулярных расстояний или малого смешения (без разрушения) узлов флуктуационной сетки. Происходит увеличение свободного объема при неизменной температуре за счет действия механических напряжений.
Схематическое изображение кривой напряжение деформация стеклообразного полимера: σт- предел текучести (предел вынужденной эластичности); звёздочкой отмечен момент разрушения.
Рис. 4. Различные стали деформации стеклообразного полимера: а- исходный образец; б-незначительное уменьшение поперечного сечения за счет увеличения общей длины; в- начало образования шейки; г- весь образец перешел в шейку; кроме тех его частей, которые находятся в зажимах динамометра; д- разрушение шейки. На рис. 4 схематически изображен внешний вид образцов на разных стадиях растяжения. Видно, что на первой стадии не происходит изменения формы образца: он удлиняется как единое целое. Деформация на первой стадии составляет доли процента или несколько процентов. Всякий образец полимера, даже внешне абсолютно однородный, имеет микродефекты структуры. Чаще всего это дефекты на поверхности, возникшие при изготовлении образца (изделия, детали и т. п.). Однако возникают и внутренние микродефекты. Они связаны либо также с процессом получения образца (частицы пыли, пузырьки воздуха и т. д.), либо с возникновением особенно крупных флуктуаций плотности, больших по размеру узлов флуктуационной сетки или больших микропустот, возникших в результате концентрирования свободного объема.
Рис. 5.концентрация напряжений при растяжении Образца, имеющего дефект в виде микротрещины. Дефекты структуры всегда являются концентраторами напряжений. Пример концентрации напряжения на микротрещине показан на рис. 5. При среднем напряжении σ величина перенапряжения σ' в вершине трещины (показано стрелками) может превышать а в десятки раз. Действующее в вершине микротрещины перенапряжение σ' приводит к вынужденному перемещению части сегментов или групп сегментов (надмолекулярных структур), расположенных в непосредственной близости от вершины. Перемещение сегментов под действием механического напряжения облегчается тем, что в образце накоплен дополнительный свободный объем при растяжении на первой стадии. Перемещение сегментов в вершине микродефекта приводит к их ориентации в направлении действия силы, материал в этом месте упрочняется, трещина не растет далее, но продолжающееся растяжение приводит к тому, что область ориентации увеличивается. Следствием ориентации является уплотнение материала, и в том месте, где произошла ориентация сегментов, на образце возникает так называемая шейка (см. рис. 4). Возникновение шейки в образце совпадает с появлением максимума на кривой σ—ε. После возникновения шейки напряжение в образце несколько снижается. Это, по-видимому, тиксотропный эффект, подобно тому как мокрый песок на берегу образует твердый малодеформируемый слой, который, однако, становится жидким и легко деформируемым, если его перемешать, разрушив существующую в нем исходную тиксотропную структуру. Одной из причин появления максимума на кривой σ—ε может быть также наличие остаточных напряжений в образце, которые реализуются в точке максимума и снижают внешнее усилие. (Иногда по аналогии с металлами максимум напряжения при σ=σт называют «зубом текучести».) Напряжение, соответствующее максимуму на кривой σ—ε, обозначают σт . При дальнейшем растяжении образца область шейки растет постепенно до тех пор, пока весь образец не перейдет в шейку (см. рис. 4). Напряжение остается при этом постоянным, и на кривой σ—ε возникает горизонтальный участок (II на рис. 3). Степень ориентации сегментов в шейке оказывается высокой. Величина деформации на стадии II достигает сотен процентов. Если образец освободить теперь из зажимов, то он, будучи застеклованным, не сократится самопроизвольно. Исчезнет только упругая деформация (доли процента). Однако при нагревании выше Тс, как только сегменты обретут вновь способность к тепловым перемещениям, образец сократится до длины, близкой к исходной. Таким образом, при растяжении стеклообразного полимера возникает ориентация сегментов в направлении действия силы, т.е. частичное разворачивание молекулярных клубков, а после нагревания выше Тс происходит свертывание макромолекул, переход их в прежнее состояние статистически свернутых клубков. Формально это явление похоже на высокоэластическую деформацию. Однако разворачивание клубков происходит вынужденно, под влиянием значительных внешних напряжений, а не в результате теплового движения. Способность стеклообразных полимеров к большим деформациям называют явлением вынужденной эластичности, а сами деформации – вынужденно - эластическими. После того как шейка сформировалась, процесс растяжения переходит в стадию III: образец растягивается как единое целое, перестройка надмолекулярной структуры теперь уже не происходит. Механизм деформации аналогичен таковому на первой стадии, но применительно к высокоориентированному образцу. Деформации составляют также несколько процентов или десятки процентов (стадия III на рис. 3). Кривая, схематически изображенная на рис. 3, является полной кривой σ—ε стеклообразного полимера. Часто полную кривую не удается получить, поскольку образец разрывается уже на второй стадии деформации. Мы видели, что перемещение сегментов в процессе вынужденно - эластической деформации происходит под действием напряжения, а не в процессе теплового перемещения. Однако определенный запас тепловой энергии в полимере имеется и при Т < Тс. С ростом температуры в области ниже Тс запас тепловой энергии сегментов увеличивается, и требуется все меньше внешней механической энергии для перемещения сегментов и развития вынужденно - эластической деформации. Поэтому предел вынужденной эластичности уменьшается с ростом Т. Формы кривой σ—ε при разных температурах приведены на рис. 6. При понижении температуры не только увеличивается предел вынужденной эластичности, но и сама кривая вырождается, становится неполной. Разрушение образца может произойти даже раньше, чем достигнут предел вынужденной эластичности σт. При этом (кривая 1 на рис. 6) разрушение происходит при очень малых деформациях (доли процента), а это означает, что полимер при низких температурах ведет себя как хрупкий, не способный к вынужденно - эластическим деформациям. Время релаксации экспоненциально зависит от абсолютной температуры: τ=τ0eU/RT (1) где U—энергия активации, т. е. тот (по аналогии с химической реакцией) потенциальный барьер, который необходимо преодолеть, чтобы осуществилось перемещение сегмента из исходного положения в соседнее, незанятое.
Т1<T2<T3<T4<T5; 1- при T1<Tхр ; 5- при T5 ≥Tc , когда деформация развивается без образования шейки. Если рассматривать деформацию стеклообразного полимера с точки зрения зависимости (1), то можно сделать вывод, что в точке стеклования энергетический барьер U на пути перемещения сегмента столь велик, что процесс этот становится невозможным. Если под действием внешнего напряжения перемещения все-таки реализуются, что приводит к вынужденно - эластической деформации, то это означает, что под действием деформирующей силы энергетический барьер стал меньше. Можно сказать, что энергия активации U является функцией напряжения. Ее можно выразить следующим образом: U= U0-aσ. Теперь можно изменить выражение (1) и получить формулу Александрова—Гуревича: τ=τ0eu◦-aσ/RT (2) В формуле (2) зафиксирована зависимость времени релаксации от напряжения. Релаксационные процессы происходят не только под влиянием теплового движения, но и под влиянием действующей силы, т. е. тогда, когда сегмент накапливает суммарный запас тепловой и механической энергии, достаточный для преодоления энергетического барьера. Рассматривая механизм вынужденно - эластической деформации, следует учитывать, что влияние механических напряжений на время релаксации является решающим лишь в момент начала образования шейки, т. е. в начале роста микротрещин. Когда перемещение сегментов в направлении деформирующего усилия становится значительным, развивается и значительное внутреннее трение. В области перехода образца в шейку (область сужения) выделяется тепло и температура повышается. Так, прямые измерения на примере полиамида показали рост температуры в области сужения на 30 ºС. Очевидно, что сам распад кристаллитов в полиамиде требовал бы, наоборот, затраты тепла, т. е. тепловой эффект обусловлен внутренним трением при вынужденно - эластической деформации. Аналогичное повышение температуры отмечено и при деформации, по существу, аморфного поливинилхлорида. Конечно, в стеклообразном состоянии вклад механической энергии является решающим; как следует из изложенного, в его отсутствие релаксационные процессы вообще не происходят. Это позволяет применять стеклообразные полимеры в качестве конструкционных материалов, изготовляя из них детали, работающие в условиях заданных деформаций или напряжений. Если стеклообразный полимер деформирован на определенную величину, меньшую, чем деформация, соответствующая пределу вынужденной эластичности, то и напряжение при этом меньше, чем σт .Релаксация напряжения при малой деформации незначительна, и напряжение, возникшее при заданной деформации, сохраняется. Образец (изделие) сохраняет размеры и формы под нагрузкой. Это отличает стеклообразные полимеры от эластомеров.
Рис. 7. Ползучесть полистирола при 25 ºС и различных значениях действующего напряжения Для стеклообразных полимеров особенно важна способность выдерживать длительное действие внешней силы (нагрузки) сохранении размеров в заданных пределах. Это определяется величиной и закономерностями ползучести. На рис. 7 показаны кривые ползучести полистирола при разных нагрузках. Видно, что при нагружении мгновенно увеличивается длина образца за счет развития упругой деформации. Далее развивается замедленная упругость, качественно аналогичная развитию высокоэластической деформации (элемент Кельвина—Фойгта). Замедленная упругость характеризует развитие вынужденно - эластической деформации. Далее возможны два случая: либо деформация перестает расти после достижения определенной величины, либо она развивается непрерывно. В первом случае мы говорим, что имеет место затухающая ползучесть, во втором случае — незатухающая ползучесть. Последняя развивается как за счет истинно необратимой, так и за счет замедленной вынужденно - эластической деформации без образования шейки. Полимер может применятся как конструкционный материал только в том случае, если под действием заданной нагрузки в нем развивается затухающая ползучесть, позволяющая обеспечить относительное постоянство размеров детали в условиях эксплуатации. Исследование поведения стеклообразных полимеров в условиях циклических деформаций позволяет обнаружить некоторые релаксационные переходы при Т≤Тс. На рис. 8 схематически показаны релаксационные переходы в полиметилметакрилате.
Рис. 8. Вторичные релаксационные переходы в полиметилметакрилате (пояснение в тексте)
Релаксационный переход, соответствующий Тс, называется главным, или α- переходом. Другие переходы — это соответственно β- и γ - переходы. Причины переходов, их молекулярный механизм не всегда можно однозначно установить. В случае полиметилметакрилата (ПММА) установлено, что при частоте внешнего деформирующего напряжения 1 Гц α-переход наблюдается при Т= Тс = 100 °С и обусловлен, как мы знаем, тем, что именно при этих условиях сегменты в полиметилметакрилате следуют за изменением вектора напряжения. На эти перемещения затрачивается много механической энергии, которая переходит в теплоту за счет внутреннего трения сегментов. При охлаждении до Т<ТС сегменты теряют подвижность и потери уменьшаются. При комнатной температуре (порядка 20 ºС) каждое изменение вектора напряжения сопровождается поворотом к (вращением) эфирной группы — СООСН3 вокруг связи С—С, соединяющей эфирную группу с главной цепью. На эти перемещения затрачивается меньше энергии, чем на перемещение сегментов, поэтому высота пика, соответствующего β - переходу, меньше, I чем высота пика α -перехода. Наконец, дальнейшее охлаждение «замораживает» и движения эфирных групп. Только в области температур около —267 ºС частота вращения метильных групп в группах — СООСНЗ. начинает совпадать с частотой поля, и мы наблюдаем γ -переход. Релаксационные переходы в стеклообразных полимерах — β, γ и т. д. — называются вторичными релаксационными переходами. Они оказывают существенное влияние на механические свойства, особенно на хрупкость и сопротивление ударным нагрузкам. ЯВЛЕНИЕ ХРУПКОСТИ ПОЛИМЕРНЫХ СТЕКОЛ Обычное оконное стекло всегда хрупко. Органическое стекло, I как мы часто называем полиметилметакрилат, менее хрупко. Его I можно уронить, не разбив. Если взять другие стеклообразные полимеры, такие как полистирол, поливинилхлорид, поликарбонат и др., то окажется, что, во-первых, они все значительно менее хрупки, чем силикатное (оконное) стекло, а во-вторых, хрупкость их очень различается. Для нас стеклообразные полимеры ценны в первую очередь тем, что они обладают пониженной хрупкостью по сравнению с силикатным стеклом, т. е. большим сопротивлением разрушению при ударе. Определим понятие хрупкости и пути ее регулирования. Хрупкость — это способность стеклообразных полимеров разрушаться при малых деформациях, меньших, чем деформация, соответствующая пределу вынужденной эластичности. На рис. 6 кривая 1 типична для хрупкого полимера. Полимер становится хрупким тогда, когда время до разрушения много меньше, чем время релаксации, и поэтому никакой перегруппировки сегментов под действием силы не происходит. Это и определяет незначительную величину деформации при разрушении. Вынужденно - эластические деформации в хрупких полимерах развиться не успевают, но вследствие наличия остаточного свободного объема стеклообразном полимере (порядка 2,5 %) происходит его хрупкое разрушение при деформации около 1 % (или немного больше), в то время как силикатные стекла разрушаются при деформации 0,1%. Хрупкость полимерных стекол принято оценивать по величине температуры хрупкости Тхр. Чем выше Тхр, тем более хрупким считается полимер. Температура хрупкости—это температура, при которой полимер разрушается в момент достижения предела вынужденной эластичности. Чтобы определить Тхр, строят зависимость предела вынужденной эластичности σт от температуры. Как следует и рис. 6, σт (максимум на кривой σ—ε) увеличивается с уменьшением температуры. Зависимость σт — Т приведена на рис. 9. Koгда температура становится ниже Тхр, вынужденная эластичность не развивается, и тогда определяют прочность полимера σр, который стал хрупким. На рис. 9 приведена также кривая зависимости σр от температуры. Точка пересечения кривых (σр = σт) и определяет Тхр. Зная Тхр и Тс, можно определить интервал температур, в котором полимер ведет себя как упругий нехрупкий материал. Если эластомеры применяют при температуре в пределах интервала высокоэластичности (между температурами стеклования и текучести), то стеклообразный полимер (пластмассу) применяют в интервале вынужденной эластичности (Тс —Тхр). Полиметилметакрилат можно применять как конструкционный материал, потому ЧЯ для него Тс = 110 ºС, а Тхр = 10 ºС. Полистирол нельзя применять без специальной модификации его структуры, потому что для него Тс =100°С, а Тхр=90°С. Температура хрупкости, как и Тс, зависит от молекулярной массы (рис. 10). При малой молекулярной массе, когда мы имеем дело с олигомером, значения Тс и Тхр совпадают. Когда молекулы становятся достаточно длинными и, следовательно, появляется гибкость, Тс растет быстрее, чем Тхр и возникает температурный интервал вынужденной эластичности (Тс—Тхр ) При дальнейшем росте молекулярной массы Тхр даже несколько
Рис. 9. Зависимость прочности σр и предела вынужденной эластичности σт от температуры понижается, что приводит к увеличению интервала вынужденной эластичности для высокомолекулярных полимеров. Из рис. 10 видно также, что с ростом молекулярной массы непрерывно ухудшается способность полимеров к необратимым деформациям. Это отражается в росте температуры текучести с ростом молекулярной массы. Рис. 10 показывает улучшение эксплуатационных характеристик полимеров вообще (эластомеров и пластмасс) с ростом молекулярной массы: растут температурные интервалы высокоэластичности (Тт —Тс) и вынужденной эластичности (Тс— Т хр).
Для ряда полимеров увеличение молекулярной массы недостаточно для обеспечения нужной протяженности температурных интервалов эластичности и вынужденной эластичности (отсутствия хрупкости). Прибегают к другим путям расширения интервалов, тем более что значительный рост молекулярной массы существенно затрудняет переработку полимеров. Эластомеры для расширения температурного интервала высокоэластичности вулканизуют. Пластмассы для снижения температуры хрупкости модифицируют. Снижению хрупкости способствует наличие в полимере таких групп атомов, которые участвуют во вторичных релаксационных переходах. Так, в полиметилметакрилате при комнатной темпера туре наблюдается широкий В-переход. Подведенная механическая энергия, например энергия удара, расходуется на повороты боковых эфирных групп в ПММА так, что рост возникающих трещин прекращается и полимер не разрушается. Вторичные релаксационные переходы, снижающие хрупкость, наблюдаются в поликарбонате, полиэтилентерефталате и других полимерах. Если вторичные релаксационные переходы отсутствуют в нужной температурной области, как, например, в полистироле, то полимер модифицируют, вводя в него эластомеры. Эластомеры образуют в хрупкой матрице полистирола множество мелких частиц, препятствующих росту трещин, возникших при ударе. Полистирол с диспергированным в нем эластомером называют «ударопрочным полистиролом», он становится хрупким лишь при значительном охлаждении. Низкомолекулярные пластификаторы, которые, как мы виде ли, снижают Тс, снижают также и Тхр. Однако Тс - при этом снижается быстрее, чем Тхр и поэтому интервал Тс –Тхр уменьшается с увеличением содержания пластификатора. Температура хрупкости определяет морозостойкость полимеров. Методы определения морозостойкости - это, как правило, методы определения той температуры, при которой полимер начинает хрупко разрушаться. Так, полимер в виде бруска, закрепленного консольно, охлаждают, определяя температуру, при которой он разрушается под действием заданного груза, падающего на него. Другой способ, применяющийся для пленочных материалов, состоит в том, что пленку сгибают в виде петли и охлаждают. Температура, при которой сплющивание петли приводит к излому пленки, характеризует морозостойкость пленки. Все методы определения морозостойкости так или иначе состоят в определении температуры, при которой полимер хрупко разрушается либо в условиях действия нагрузки заданной величины, либо деформирования на заданную величину. Методы определения морозостойкости имеют прикладное значение и приводятся в соответствующих ГОСТах. Температура, характеризующая морозостойкость, сильно зависит от метода ее определения и обычно не совпадает с Тхр, определенной так, как показано на рис. 9. Итак, при охлаждении полимеров до Т = Тс свободный объем становится недостаточным для теплового перемещения сегментов. Это проявляется в потере полимером эластичности или способности к самопроизвольному сокращению после деформации. Поскольку время релаксации уменьшается под действием механического напряжения, сегменты сохраняют способность к перемещению под действием внешней силы без разрушения полимера. Наблюдающаяся при этом значительная вынужденно - эластическая деформация не исчезает в стеклообразном полимере после снятия нагрузки, хотя и обусловлена развертыванием молекулярных клубков под действием внешнего деформирующего; усилия Охлаждение полимера до температуры ниже Тс может привести и к потере способности к вынужденно - эластической деформации — полимер перейдет в хрупкое состояние. Существенно важной чертой полимерных стекол является то, что при Т< Тс в них самопроизвольно происходят релаксационные переходы, связанные с переремещением молекулярных группировок, меньших, чем размер сегмента. Это привадит к диссипации энергии, в том числе энергии удара, и делает полимерные стекла существенно более стойкими к удару по сравнению с низкомолекулярными силикатными стеклами. Список использованной литературы. 1) В. Н., Кулезнёв, В. А. Шершнёв «Химия и физика полимеров».-2-е издание, перераб. и доп.- Москва «КолосС» 2007 г. 2) И. И. Тугов, Г. И. Кострыкина «Химия и физика полимеров». Москва «Химия» 1989 3) А. А. Тагер «Физика-химия полимеров».-2-е издание. Москва «Химия» 1988 г. 4) Г. М. Бартенев, С. Я Френкель «Физика полимеров» Ленинград «Химия» 1990 г. 5) Г. М.Бартенев, Ю. В. Зеленев «Курс физики полимеров» Москва 1976 г. 6) В. Е. Гуль; В.Н. Кулезнев. «Структура и механические свойства полимеров» Москва «Высшая школа», 1966 г. 7) И. П. Антонова, Антипова «Химия и физика полимеров Москва, 2001 8) Авторы: В.Ф. Куренков, Н.И. Авакумова, Л.А. Бударина. «Практикум по химии и физике полимеров» Москва, 1990 г. |
 Как
известно, полимер, находящийся в высокоэластическом состоянии, может потерять
способность к большим обратимым деформациям, если уменьшить продолжительность
действия силы.
Как
известно, полимер, находящийся в высокоэластическом состоянии, может потерять
способность к большим обратимым деформациям, если уменьшить продолжительность
действия силы.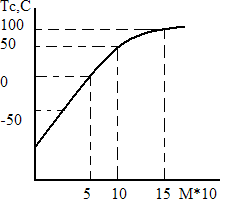
 Влияние состава
сополимера. Величина Тс меняется монотонно с изменением состава
сополимера в соответствии с уравнением Флори—Фокса
Влияние состава
сополимера. Величина Тс меняется монотонно с изменением состава
сополимера в соответствии с уравнением Флори—Фокса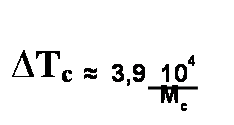
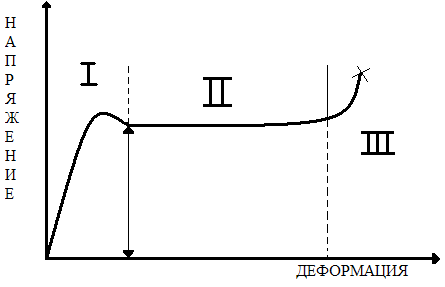 Рис 3.
Рис 3.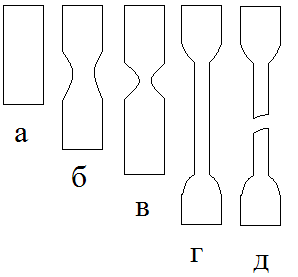
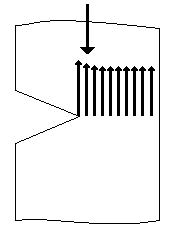
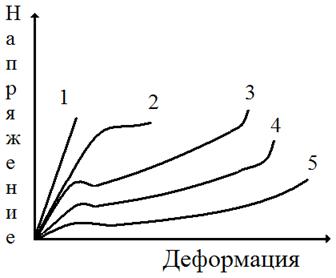 Рис. 6.
Влияние температуры на вид кривых напряжение – деформация для стеклообразных
полимеров:
Рис. 6.
Влияние температуры на вид кривых напряжение – деформация для стеклообразных
полимеров: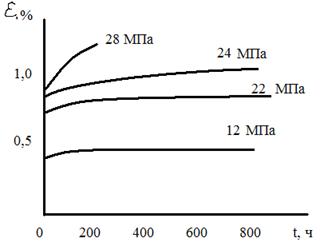
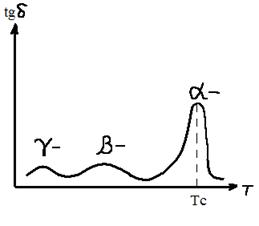
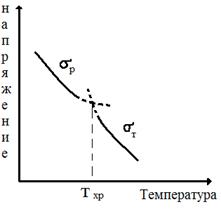
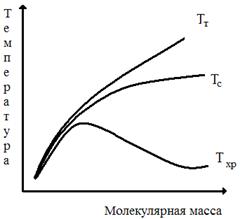 Рис.
10. Зависимость температур текучести (Т т ), стеклования (Тс)
и хрупкости (Т хр) от молекулярной массы полимера
Рис.
10. Зависимость температур текучести (Т т ), стеклования (Тс)
и хрупкости (Т хр) от молекулярной массы полимера