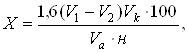Контрольная работа: Методы исследование свойств сырья и продуктов питанияКонтрольная работа: Методы исследование свойств сырья и продуктов питанияМетрологические основы контроля качества исследовательских работ. Типы погрешности. Метрологические характеристики методов и методикВзаимосвязь качества и измерений неразрывна, но ведущим является качество, именно для его обеспечения требуются измерения. Изменения в подходах к обеспечению качества, к управлению качеством в значительной степени влияют на метрологическую деятельность на предприятии. Увидеть, воспринять, принять соответствующие научно-технические и организационные решения для адаптации метрологической деятельности, значит сделать метрологию в исследовательской работе эффективной. Под метрологическим обеспечением измерений понимается установление и применение научных и организационных основ, технических средств, правил и норм, необходимых для достижения единства и требуемой точности измерений. Понятие «метрологическое обеспечение» применяется, как правило, по отношению к измерениям, испытанию и контролю в целом. В то же время допускается использование понятия метрологическое обеспечение (МО) технологического производства. При этом МО может включать в себя различные этапы технологических процессов, например: – установление рациональной номенклатуры измеряемых параметров и оптимальных норм точности измерений; – технико-экономическое обоснование и выбор средств измерений (СИ), испытаний и контроля; – стандартизация, унификация и агрегатирование используемой контрольно-измерительной техники; – разработка, внедрение и аттестация современных методик выполнения измерений, испытаний, контроля; – поверка, метрологическая аттестация и калибровка контрольно-измерительного и испытательного оборудования; – контроль за производством, состоянием и использованием КИА; – разработка и внедрение стандарта предприятия; – внедрение международных и отраслевых стандартов; – проведение метрологической экспертизы проектов; – проведение анализа состояния измерений и т.п. МО в исследовательской работе имеет четыре основы: научную, организационную, нормативную, и техническую. Организационной основой метрологического обеспечения является метрологическая служба РФ, основным ее звеном является государственная метрологическая служба (ГМС), возглавляемая госкомитетом по стандартам (Госстандартом). Госстандарт руководит разработкой научно-методических, технико-экономических, правовых и организационных основ метрологического обеспечения. В областях, где надзор и контроль не применяются, используются правила и положения, введенные положением Российской системы калибровки. Научно-технические проблемы метрологического обеспечения решаются в метрологических НИИ, организационные проблемы (практическая метрология) решаются в основном территориальными организациями Госстандарта. Метрологическая деятельность в РФ основывается на конституционной норме (ст. 71). В развитие этой конституционной нормы приняты законы «Об обеспечении единства измерений» и «О стандартизации», детализирующие основы метрологической деятельности. Мерой оценки точности измерений является погрешность. Погрешность характеризует отклонение измеренного значения некоторой величины от её истинного (действительного) значения. Следует различать погрешность измерений, получаемую как результат обработки экспериментальных наблюдений, и нормированную погрешность средства измерения, являющуюся его технической характеристикой. Эти погрешности могут совпадать только в отдельных частных случаях. Абсолютной погрешностью называется разность D = x – X, где х – истинное значение; Х – результат измерения. Абсолютная погрешность выражается в тех же единицах, что и измеряемая величина. Относительной погрешностью измерения называется отношение метрологический вольтамперметрический пищевой теплоемкость
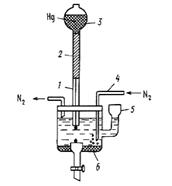
По своей природе погрешности бывают систематическими и случайными. Систематическая погрешность – составляющая погрешности результата измерения, остающаяся постоянной или закономерно изменяющаяся при повторных измерениях одной и той же физической величины. Систематические погрешности, которые действуют в процессе измерения, называются неисключёнными. Классификация систематических погрешностей: 1 – инструментальные, которые свойственны средствам измерения и являются следствием дефектов их статических характеристик; 2 – методические, возникающие из-за несовершенства методики измерения либо из-за несоответствия методики поставленной задаче; 3 – субъективные, вызванные ошибками наблюдателя при отсчёте показаний (небрежность, параллакс, ошибка при интерполяции). Нормированная погрешность является паспортной характеристикой средства измерения и может быть задана в виде абсолютной, относительной или приведенной погрешности. В некоторых случаях, например при изготовлении нестандартных средств или при желании сузить пределы допустимой систематической погрешности, их подвергают индивидуальным градуировкам. Пределы допустимых погрешностей средств измерений (паспортные или индивидуальные) должны рассматриваться как границы основной неисключенной систематической погрешности. Уменьшить систематические погрешности можно, устраняя или уменьшая изменения внешних условий (стабилизация питающего напряжения, термостабилизация, экранирование и т.п.) Единственным путём выявления необнаруженных систематических погрешностей является проведение измерений двумя или несколькими независимыми методами, обладающими приблизительно одинаковыми постоянными и переменными систематическими погрешностями. Грубое расхождение между результатами, полученными разными методами, указывает на наличие в одном из каналов измерений недопустимой систематической погрешности. Случайная погрешность – составляющая погрешности результата измерения, изменяющаяся случайным образом (по знаку и значению) при повторных измерениях, проведенных с одинаковой тщательностью, одной и той же физической величины. Методы теории вероятностей и математической статистики позволяют установить вероятностные (статистические) закономерности появления случайных погрешностей и на основании этих закономерностей дать количественные оценки результата измерений и его случайной погрешности. Критериями для оценки и выбора методов контроля служат их метрологические характеристики (интервал определяемых содержаний, верхняя и нижняя границы определяемого содержания веществ, предел обнаружения (чувствительность), воспроизводимость, правильность), а также аналитические характеристики (селективность, продолжительность, производительность. Интервал определяемого содержания веществ – это предусмотренная данной методикой область значений определяемых содержаний веществ. Физические, химические и физико-химические методы исследования применяют для количественного определения веществ в широких пределах относительных содержаний: основных (100–1%); неосновных (1,0–0,01%); следовых (меньше 0,01%), а также содержания частей определяемого компонента: на миллион частей основы lppm = 1 х 10–4% или на миллиард частей основы lppb = – 1 х 10–7%. Нижняя граница определяемых содержаний (Сн) – это наименьшее значение определяемого содержания, ограничивающее интервал определяемых содержаний. Верхняя граница определяемых содержаний (Св) – это наибольшее значение определяемого содержания, ограничивающее интервал определяемых содержаний. (Сн) и (Св) обычно представляют собой массовую долю определяемого компонента в исследуемом продукте, а не в растворе. Предел обнаружения (Смин) – наименьшее содержание, при котором по данной методике можно обнаружить присутствие определяемого компонента с заданной доверительной вероятностью. Метод или методика анализа дают лишь тогда правильный результат, когда он свободен от систематических погрешностей. Систематические погрешности могут возникать на любом этапе аналитического процесса и по разным причинам. Задача освобождения результатов измерения от систематических погрешностей требует глубокого анализа всей совокупности данных измерений. Например, наиболее вероятным источником систематических погрешностей фотометрических измерений могут служить недостаточная представительность состава отобранной аналитической навески, погрешности в подготовке аналитической навески к фотометрическим измерениям, погрешности градуировки весов, мерной посуды, шкал спектрофотометров, несоответствие составов анализируемых и стандартных растворов, по которым строились градуировочные графики. Одной из часто встречающихся в физико-химических методах анализа причин систематических погрешностей является неправильное градуирование, в частности, построение градуировочных графиков на основе неподходящих градуировочных проб. Воспроизводимость и сходимость результатов анализа определяются разбросом повторных результатов анализа относительно их среднего значения и обусловливаются наличием случайных погрешностей. Сходимость характеризует рассеяние результатов при фиксированных условиях выполнения эксперимента; воспроизводимость – при варьировании этих условий. В первом приближении можно считать, что стандартное отклонение сходимости в 1,4–1,5 раза меньше стандартного отклонения внутри лабораторной воспроизводимости. Ввиду наличия такой простой связи между характеристиками сходимости и воспроизводимости в дальнейшем будет идти речь лишь о воспроизводимости как более общепринятом в литературе понятии. Чем больше значение аналитических и инструментальных случайных погрешностей, тем менее точен анализ. Воспроизводимость характеризуется значением стандартного отклонения (S) или относительного стандартного отклонения (Sr). Вольтамперметрические методы анализа пищевых продуктовМетоды анализа, основанные на расшифровке поляризационных кривых (вольтамперограмм), получаемых в электролитической ячейке с поляризующимся индикаторным электродом и неполяризующимся электродом сравнения, называют вольтамперометрическим. Вольтамперограмма позволяет одновременно получить качественную и количественную информацию о веществах, восстанавливающихся или окисляющихся на микроэлектроде (деполяризаторах), а также о характере электродного процесса. В качестве поляризующегося микроэлектрода часто применяют ртутный капельный электрод, а сам метод называют в этом случае полярографией, следуя термину, который предложил Я. Гейровский, разработавший этот метод в 1922 г. При небольшом потенциале катода сила тока сначала медленно увеличивается с возрастанием потенциала – это так называемый остаточный ток, его значение имеет порядок 10-7 А. По достижении потенциала восстановления на катоде начинается разряд ионов, определяемый диффузией, и сила тока резко возрастает, а затем становится постоянной – это предельный диффузионный ток. Принципиальная схема полярографической установки приведена на рисунке 1. Полярографическая установка включает в себя резервуар с ртутью, соединённый шлангом с капилляром, погруженным в анализируемый раствор. Электродом сравнения может служить слой донной ртути. В настоящее время, однако, чаще применяют обычные электроды сравнения – каломельный или хлоридсеребряный.
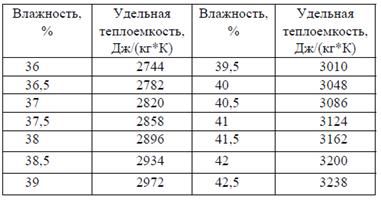
Рис. 1. Простейшая полярографическая ячейка: 1 – стеклянный капилляр; 2 – полиэтиленовый шланг; 3 – груша с металлической ртутью; 4 – стеклянная трубочка с оттянутым концом для ввода азота; 5 – воронка для смены раствора; 6 – донная ртуть (Hg-анод) Зависимость тока I от приложенного напряжения Е при обратимом электродном процессе передается уравнением полярографической волны: Е = Е1/2 + (R T / n F) ln (Id – I) / I, (1) Где Е1/2 – потенциал полуволны; Id – диффузионный ток. При I = Id / 2 уравнение (1) переходит в Е = Е1/2. (2) Это соотношение показывает независимость потенциала полуволны от тока и, следовательно, от концентрации восстанавливающегося иона. Потенциал полуволны является, таким образом, качественной характеристикой иона в растворе данного фонового электролита, и определение потенциала полуволны составляет основу качественного полярографического анализа. Количественный полярографический анализ основан на уравнении Ильковича, которое связывает диффузионный ток Id с концентрацией иона с и рядом других величин: Id = 605 z D1/2 m 2/3 t1/6 c (3) где z – заряд иона; D – коэффициент диффузии; m – масса ртути, вытекающей из капилляра за 1 с, мг; t – время образования капли (периода капания), с. В практике количественного полярографического анализа коэффициент пропорциональности межу концентрацией вещества и силой диффузионного тока обычно устанавливают с помощью стандартных растворов. При постоянных условиях полярографирования D, m, и t постоянны, поэтому уравнение (3) переходит в Id = k c. (4) При анализе некоторых систем, для которых применимость уравнения (4) установлена вполне надежно, часто используют менее трудоемкий метод стандартных растворов. Так же широко распространен в количественной полярографии и метод добавок. Особое место в полярографическом анализе занимает амперометрическое титрование. Амперометрическое титрование представляет собой разновидность полярографического метода анализа. Амперометрическое титрование проводится следующим образом: часть исследуемого раствора помещают в электролизер, снабженный индикаторным электродом и электродом сравнения. Между электродами устанавливают напряжение на 0,3 – 0,5 В больше потенциала полуволны (или редокс-потенциала) исследуемого вещества и приступают к титрованию. В процессе титрования отмечают показания гальванометра, на основании результатов строят кривую амперометрического титрования, откладывая на оси ординат показания гальванометра, а на оси абсцисс – объем титранта. Точка перегиба соответствует объему титранта в точке эквивалентности. Содержание определяемого вещества вычисляют по объему титранта, израсходованному в точке эквивалентности. Концентрация титранта должна превышать концентрацию раствора титруемого вещества в 10–15 раз. При амперометрическом титровании индикаторными электродами могут быть ртутный капельный электрод, платиновый вращающийся и другие электроды. В качестве электродов сравнения применяют насыщенный каломельный, хлорсеребряный и другие электроды. Вид кривой амперометрического титрования будет зависеть от того, какой компонент реакции титрования вступает в электродную реакцию и при каком потенциале ведется титрование. Сама реакция титрования, естественно, будет протекать независимо от этих условий. Амперометрическое титрование следует проводить при потенциале, отвечающем области диффузионного тока. Обычно титруют при потенциале на 0,2–0,3 В более отрицательном, чем потенциал полуволны полярографически активного соединения. Полярографическая установка служит для получения полярограмм, т.е. кривых зависимости силы тока, протекающего через раствор, от потенциала, приложенного к рабочему электроду. Прибор состоит из трех основных узлов: электролитической ячейки с рабочим электродом и электродом сравнения, источника напряжения для поляризации рабочего электрода и устройства для регистрации тока. В качестве неполяризующегося электрода сравнения используется слой ртути на дне ячейки. Применяются также и другие электроды сравнения: каломельный, ртутно-сульфатный, хлорсеребряный и др. Рабочим электродом может быть также твердый микроэлектрод, изготавливаемый из платины, золота, графита и других материалов. Установка для амперометрического титрования может быть собрана на основе любого полярографа. Обычно для этой цели используется самая простая полярографическая установка. При этом рабочим может быть как ртутный капающий, так и твердый микроэлектрод. В качестве источников тока могут применяться аккумуляторные батареи и различные выпрямительные устройства. В комплект установки для титрования входят также микробюретка и магнитная мешалка. На российском рынке аналитического оборудования представлено порядка 10 вольтамперометрических анализаторов, как правило, со своими электродами, программным и методическим обеспечением. Информацию о них можно найти в каталогах фирм-изготовителей, проспектах и буклетах различных выставочных мероприятий, в международной компьютерной сети Интернет. Практически все вольтамперометрические анализаторы управляются программным способом с помощью компьютера. Это позволяет полностью автоматизировать настройку прибора и анализ, исключить промахи, гибко и оперативно расширять функциональные возможности приборов и улучшать параметры, как уже находящихся в эксплуатации, так и новых. Вычислить теплоемкость теста при значении его влажности 39,81%Пусть Сm – удельная теплоемкость теста, Дж/(кг К) (зависит от влажности теста и определяется по таблице 1. Таблица 1. Удельная теплоемкость макаронного теста в зависимости от его влажности
Методом линейной интерполяции находим, что при влажности 39,81% Cm = 3010+(3048–3010)*(39,81–39,5) / (40–39,5) = 3034 Дж/(кг К). Титриметрический метод определения крахмала. Реакции. Расчет результатов анализаГидролиз полисахаридов является стадией предшествующей дальнейшему их анализу. Гидролиз крахмала под действием кислот вызывает ослабление и разрыв ассоциативных связей между макромолекулами амилазы и амилопектина. Далее идет разрыв α-Д – (1,4) – и α-Д – (1,6) связей с присоединением по месту разрыва молекулы воды. По мере гидролиза происходит нарастание редуцирующих сахаров. Конечным продуктом является глюкоза. К недостатку этого метода следует отнести возможность образования продуктов термической деградации и дегидратации углеводов и реакции трансгликозилирования: фурфурол, оксиметилфурфурол, 2-гидроксиацетилфуран, изомальтол, левулиновой, муравьиной, молочной, уксусной кислот и ряд других соединений. Крахмал гидролизируется и под действием амилолитических ферментов: α- и β – амилаза, глюкоамилаза, пуллуланаза и др. Для определения большого множества углеводных соединений и продуктов превращений используют: гравиметрические, титриметрические и физико-химические методы анализа (оптические, электрохимические, хроматографические и др.). HOH2C O CHO
HOH2C OH фурфурола Для определения моносахаридов используют их восстанавливающую способность. Вначале их извлекают из пищевых продуктов 80%-ным этиловым спиртом. Спиртные экстракты упаривают под вакуумом разбавляют горячей водой и фильтруют. При анализе продуктов, относительно богатых белками и фенольными соединениями, фильтрат дополнительно обрабатывают нейтральным раствором ацетата свинца, избыток удаляют сульфатом, фосфатом или оксалатом натрия. Осадок отфильтровывают, а в фильтрате определяют редуцирующие сахара титриметрическим, амперометрическим, хроматографическим, рефрактометрическим и др. методами анализа. При титриметрическом методе анализа к аликвотной части экстракта прибавляют 25 см3 щелочного раствора гексацианоферрата (III) калия, нагревают и титруют раствором глюкозы в присутствии индикатора метилового голубого до исчезновения синей окраски. Содержание (%) определяют по формуле:
где – V1 – количество стандартного раствора глюкозы, пошедшее на титрование 25 см3 щелочного раствора гексацианоферрата (III) калия, см3; V2 – количество стандартного раствора глюкозы, пошедшее на титрование избытка гексацианоферрата (III) калия, см3; Vк – объем экстракта, см3; Vа – аликвотная часть экстракта, см3; 1,6 – масса глюкозы в 1 см3 стандартного раствора, мг; н – навеска объекта исследования, мг. Список литературы1. Донченко Л.В., Надыкта В.Д. Безопасность пищевой продукции: Учебник. 2-е изд., перераб. и доп. – М.: Дели принт, 2005. 2. Методы анализа пищевых продуктов. Под редакцией Ю.А. Клячко, С.М. Беленького. – М.: Наука, 1988. 3. Роева Н.Н., Касьяненко Г.Р., Кирничная В.К. Методы исследования свойств сырья и продуктов питания. Учебно-практическое пособие. – М., МГУТУ, 2004. 4. Руководство по методам анализа качества и безопасности пищевых продуктов. Под.ред. И.М. Скурихина, В.А. Тутельмана. М.: Брандес – Медицина, 1998. 5. Химический состав пищевых продуктов. Под. ред. И.М. Скурихина, М.Н. Волгарева. М.: Агропромиздат, 1987. |
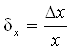 , или в процентах
, или в процентах 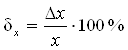
 СНО
СНО
 СН О HOH2C CHO
СН О HOH2C CHO